20190909_Count3ssRatioExample
Ben Fair
9/10/2019
Last updated: 2019-09-18
Checks: 6 1
Knit directory: cheRNA_pilot/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.4.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190813) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Unstaged changes:
Modified: analysis/20190909_Count3ssRatioExample.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 3d9467f | Benjmain Fair | 2019-09-18 | update 3ss classification |
| html | 3d9467f | Benjmain Fair | 2019-09-18 | update 3ss classification |
| Rmd | 46e6b59 | Benjmain Fair | 2019-09-16 | update site |
| html | 46e6b59 | Benjmain Fair | 2019-09-16 | update site |
| Rmd | b3b13e6 | Benjmain Fair | 2019-09-16 | update site |
| html | b3b13e6 | Benjmain Fair | 2019-09-16 | update site |
| Rmd | 4ea43f8 | Benjmain Fair | 2019-09-16 | update 3ss quantification analyses |
| html | 4ea43f8 | Benjmain Fair | 2019-09-16 | update 3ss quantification analyses |
| Rmd | 78712fa | Benjmain Fair | 2019-09-16 | update site |
| html | 78712fa | Benjmain Fair | 2019-09-16 | update site |
library(tidyverse)For each 3’ss, I used bedtools to count the number of reads overlapping the upstream 25bp window, and the downstream 25bp window. The ratio of these of these two counts is a measure of splicing efficiency, and the ratio of those ratios in nascent RNA-seq versus standard RNA-seq is a measure of how contranscriptional a splicing event is (see Herzel et al). Here I will look at these metrics using a standard RNA-seq dataset and nascent RNA-seq dataset from the same LCL line 18862.
I have already calculated counts in upstream and downstream windows for each annotated 3’ss using bedtools (see Snakemake code). Here I will investigate the results with a few plots. One result I expect to see is that alernative 3’ss will be spliced less contranscripionally. In the case of cassette exons, it is important to distinguish between the upstream 3’ss and the downstream 3’ss. Upstream 3’ss introns will naturally have less coverage in the downstream window (i.e. in the cassette exon).
First read in Gencode annotated introns and classify the 3’ss as alternative upstream, alternative downstream, or constitutive, based on how many introns are annotated with each 3’ss. This Gencode file of introns was made using gtf2leafcutter.pl script in leafcutter github repo, and it is a redundant file of all intron segments from Gencode gtf. In other words, there are many overlapping introns, including many with the same 3’ss. So here is the pseudo-code logic on how I classify introns:
Constitutive introns:
- 3’ss is paired to one and only one 5’ss in the annotations. The 5’ss that it is paired to is paired only to that 3’ss and no other 3’ss.
Downstream alternative introns (green):
- 3’ss is paired to multiple 5’ss in the annotations, and all of the 5’ss that it is paired to are only paired to that 3’ss. This will also include 3’ss that are associated with alt 5’ss usage.
Upstream alternative introns (blue):
- 3’ss is paired to a 5’ss that is used in more than 1 intron. This will also include 3’ss that are associated with alt 3’ss usage.
GencodeIntrons <- read.table("../data/GencodeHg38_all_introns.corrected.bed.gz", sep='\t', col.names = c('chrom', 'start', 'stop', 'name', 'score', 'strand', 'gene', 'intronNumber', 'transcriptType'))
SpliceSitesCounted <- GencodeIntrons %>%
filter(transcriptType=="protein_coding") %>%
distinct(chrom, start, stop, .keep_all = T) %>%
mutate(Acceptor = case_when(strand == "+" ~ paste(chrom, stop, strand, sep="."),
strand == "-" ~ paste(chrom, start, strand, sep="."))) %>%
mutate(Donor = case_when(strand == "+" ~ paste(chrom, start, strand, sep="."),
strand == "-" ~ paste(chrom, stop, strand, sep="."))) %>%
add_count(Acceptor, name="AcceptorCount") %>%
add_count(Donor, name="DonorCount")
#AcceptorCount is number of intron segments (unique Donor/Acceptor pairs) with that Acceptor 3'ss
#DonorCount is number of intron segments (unique Donor/Acceptor pairs) with that Donor 5'ss
#DonorMax is for each acceptor, what is the maximum DonorCount amongst all the donors it is paired with
Annotated <- SpliceSitesCounted %>%
# filter(AcceptorCount==1 & DonorCount==1) %>% dim()
group_by(Acceptor) %>%
mutate(DonorMax=max(DonorCount)) %>%
ungroup() %>%
distinct(Acceptor, .keep_all = T) %>%
mutate(Type3ss = case_when(AcceptorCount == 1 & DonorMax==1 ~ "Constitutive intron",
AcceptorCount >1 & DonorMax > 1 ~ "Downstream alternative intron",
AcceptorCount == 1 & DonorMax > 1 ~ "Upstream alternative intron",
AcceptorCount >1 & DonorMax==1 ~ "Unclassifiable"))
#By the above logic, "Unclassifiable"" means a 3ss that is associated with alt 5ss but not exon skipping, since none of the Donors it uses is used more than once.
table(Annotated$Type3ss)
Constitutive intron Downstream alternative intron
149839 20115
Unclassifiable Upstream alternative intron
7003 31735 # Write out as bed file to visually inspect classifications in IGV to check for bugs
# Manual inspection of the resulting bed file indiciates I am classifying introns as intended.
write.table_with_header <- function(x, file, header, ...){
cat(header, '\n', file = file)
write.table(x, file, append = T, ...)
}
Annotated %>%
mutate(NewName=paste(Acceptor, Type3ss, sep=".")) %>%
# mutate(NewStart = case_when(strand == "+" ~ stop-25,
# strand == "-" ~ start)) %>%
# mutate(NewStop = case_when(strand == "+" ~ stop,
# strand == "-" ~ start-25)) %>%
# select(chrom, NewStart, NewStop, paste(Acceptor, Type3ss), ".", strand) %>% head()
mutate(Color = case_when(Type3ss == "Constitutive intron" ~ "255,0,0",
Type3ss == "Downstream alternative intron" ~ "0,204,0",
Type3ss == "Upstream alternative intron" ~ "0,0,255",
Type3ss == "Unclassifiable" ~ "224,224,224")) %>%
mutate(thickStart=start, thickEnd=stop, newScore=0) %>%
select(chrom, start, stop, Acceptor, newScore, strand, thickStart, thickEnd, Color) %>%
# write.table("~/Temporary/3ssAnnotations.bed", quote=F, sep='\t', col.names=F, row.names = F)
write.table_with_header("~/Temporary/3ssAnnotations.bed", 'browser hide all\ntrack name="ItemRGBDemo" description="Item RGB demonstration" visibility=2 itemRgb="On"', quote=F, sep='\t', col.names=F, row.names = F)Now, similarly to Herzel et al, set a minimum read count threshold cutoff for reliable 3’ss splice ratios to use for downstream analysis. Then do some plotting. More precisely, I will plot Intron retenetion percent spliced in (Intron retention PSI) which will be \(IntronRetentionPSI=\frac{UpstreamCoverage}{DownstreamCoverage}\).
Cutoff <- 50
RS <- read.table("../output/3ssCoverageBeds/NA18862_argonne.bed.gz", sep='\t', col.names = c('chrom', 'start', 'stop', 'name', 'score', 'strand', 'upstream', 'downstream'), stringsAsFactors = F) %>% filter(upstream + downstream >= Cutoff)
nRS <- read.table("../output/3ssCoverageBeds/18862_cheRNA_1.bed.gz", sep='\t', col.names = c('chrom', 'start', 'stop', 'name', 'score', 'strand', 'upstream', 'downstream'), stringsAsFactors = F) %>% filter(upstream + downstream >= Cutoff)
ToPlot <- RS %>%
left_join(nRS, by="name") %>%
inner_join((Annotated %>% filter(Type3ss != "Unclassifiable")), by=c("name" = "Acceptor"))%>%
mutate(RS.ratio = upstream.x/(downstream.x)) %>%
mutate(nRS.ratio = upstream.y/(downstream.y)) %>%
mutate(NormalizedRatio = nRS.ratio/RS.ratio)
# Plot Intron retenetion PSI.
ggplot(ToPlot, aes(color=Type3ss)) +
stat_ecdf(aes(x=nRS.ratio, linetype="nascent-RNA"), geom = "step") +
stat_ecdf(aes(x=RS.ratio, linetype="polyA-RNA"), geom = "step") +
ylab('Cumulative fraction') +
xlab("Intron retention PSI\nMore spliced<----------->More unspliced") +
scale_x_continuous(trans='log10', limits=c(0.001,1)) +
theme_bw()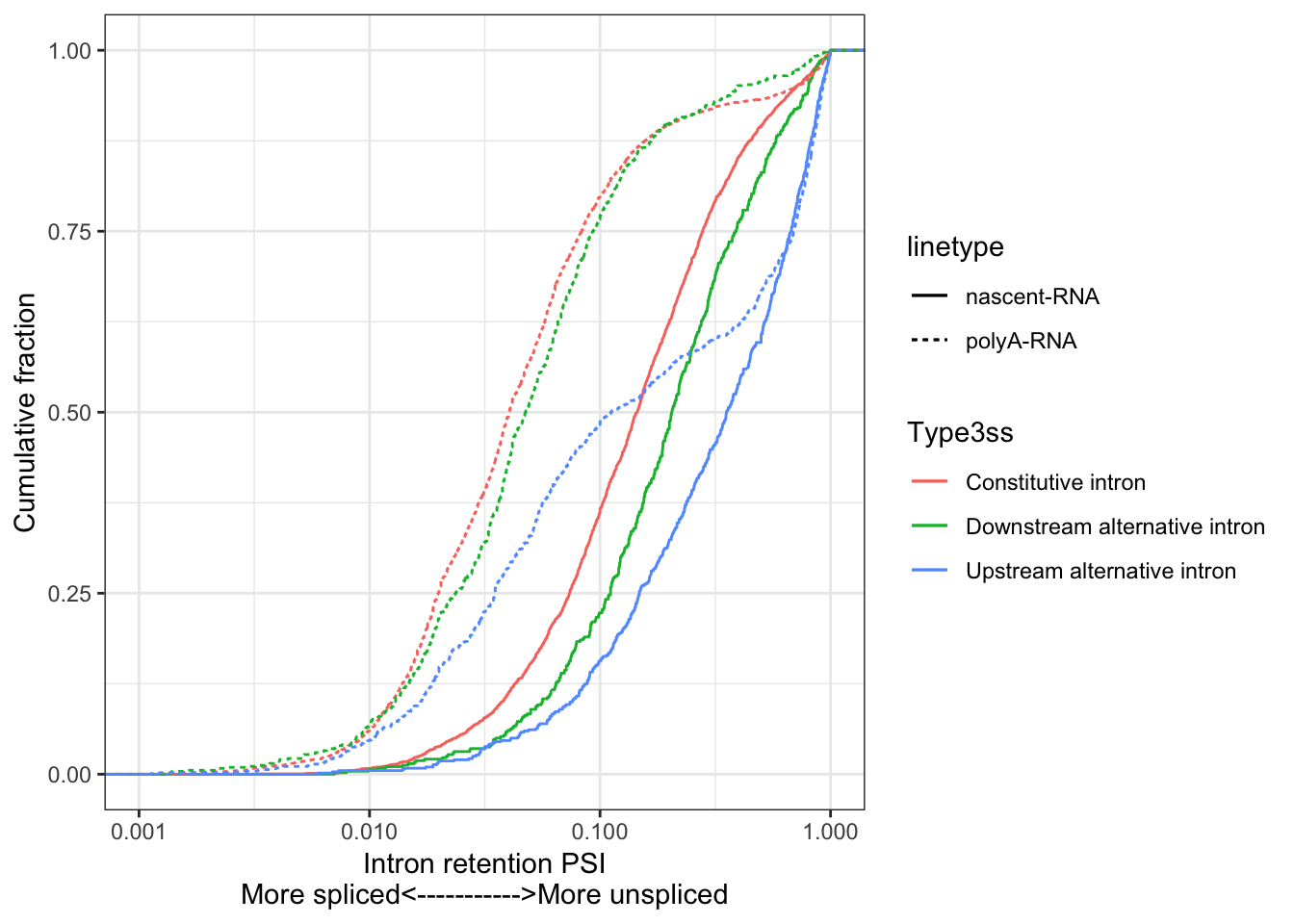
Chromatin associated RNA has higher \(Intron Retention PSI\) as expected. Alternative upstream introns also have \(Intron Retention PSI\) as expected, since there should be less coverage downstream of those 3’ss for cassette exons. Now quantify cotrnanscriptional splicing with a metric defined as \(\frac{Intron retention PSI_{nascent}}{Intron retention PSI_{polyA}}\).
# Plot cotranscriptional splicing
ggplot(ToPlot, aes(x=NormalizedRatio, color=Type3ss)) +
stat_ecdf(geom = "step") +
ylab('Cumulative fraction') +
xlab("Cotranscriptional splicing ratio\nMoreCoTxnSplicing<---------->LessConTxnSplicing") +
scale_x_continuous(trans='log10') +
theme_bw()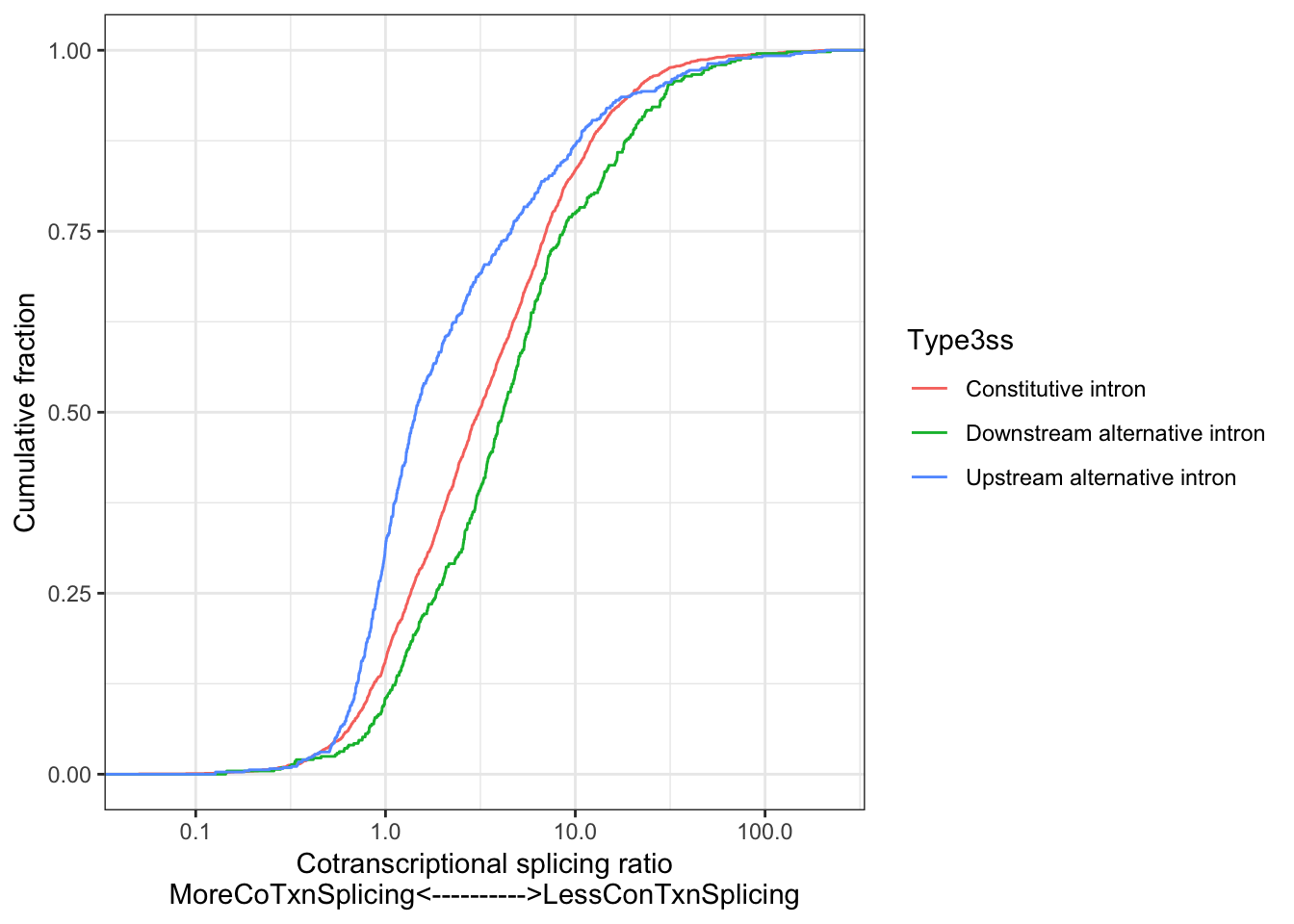
ToTest <- ToPlot %>%
filter(Type3ss %in% c("Constitutive intron", "Downstream alternative intron"))
wilcox.test(NormalizedRatio ~ Type3ss, data=ToTest)
Wilcoxon rank sum test with continuity correction
data: NormalizedRatio by Type3ss
W = 459050, p-value = 8.632e-07
alternative hypothesis: true location shift is not equal to 0As expected from Herzel et al, 3’ss downstream of cassette exons (which are an intron-centric measure of splicing of the upstream cassette exon) are spliced slower than constitutive introns.
Next I should see how replicates compare, to see if this statistic is reasonably stable (eventually I could consider QTL mapping using this statistic).
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.14
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] forcats_0.4.0 stringr_1.4.0 dplyr_0.8.1 purrr_0.3.2
[5] readr_1.3.1 tidyr_0.8.3 tibble_2.1.3 ggplot2_3.1.1
[9] tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] Rcpp_1.0.1 cellranger_1.1.0 plyr_1.8.4 pillar_1.4.1
[5] compiler_3.5.1 git2r_0.25.2 workflowr_1.4.0 tools_3.5.1
[9] digest_0.6.19 lubridate_1.7.4 jsonlite_1.6 evaluate_0.14
[13] nlme_3.1-140 gtable_0.3.0 lattice_0.20-38 pkgconfig_2.0.2
[17] rlang_0.3.4 cli_1.1.0 rstudioapi_0.10 yaml_2.2.0
[21] haven_2.1.0 xfun_0.7 withr_2.1.2 xml2_1.2.0
[25] httr_1.4.0 knitr_1.23 hms_0.4.2 generics_0.0.2
[29] fs_1.3.1 rprojroot_1.3-2 grid_3.5.1 tidyselect_0.2.5
[33] glue_1.3.1 R6_2.4.0 readxl_1.3.1 rmarkdown_1.13
[37] modelr_0.1.4 magrittr_1.5 whisker_0.3-2 backports_1.1.4
[41] scales_1.0.0 htmltools_0.3.6 rvest_0.3.4 assertthat_0.2.1
[45] colorspace_1.4-1 labeling_0.3 stringi_1.4.3 lazyeval_0.2.2
[49] munsell_0.5.0 broom_0.5.2 crayon_1.3.4