2024-01-17_CheckYangsJunctionAnnotator
2024-01-17
Last updated: 2024-06-06
Checks: 5 2
Knit directory:
ChromatinSplicingQTLs/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20191126) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /project2/yangili1/bjf79/ChromatinSplicingQTLs/data/igsr_samples.tsv.gz | ../data/igsr_samples.tsv.gz |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 209c2d5. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.Rhistory
Ignored: code/.DS_Store
Ignored: code/.RData
Ignored: code/._report.html
Ignored: code/.ipynb_checkpoints/
Ignored: code/.snakemake/
Ignored: code/APA_Processing/
Ignored: code/Alignments/
Ignored: code/ChromHMM/
Ignored: code/ENCODE/
Ignored: code/ExpressionAnalysis/
Ignored: code/ExtractPhenotypeBedByGenotype.py
Ignored: code/FastqFastp/
Ignored: code/FastqFastpSE/
Ignored: code/FastqSE/
Ignored: code/FineMapping/
Ignored: code/GTEx/
Ignored: code/Gencode.v34.6Colors.bed.gz
Ignored: code/Genotypes/
Ignored: code/H3K36me3_CutAndTag.pdf
Ignored: code/IntronSlopes/
Ignored: code/LR.bed
Ignored: code/LR.seq.bed
Ignored: code/LongReads/
Ignored: code/MYB.tracks.ini
Ignored: code/Metaplots/
Ignored: code/Misc/
Ignored: code/MiscCountTables/
Ignored: code/Multiqc/
Ignored: code/Multiqc_chRNA/
Ignored: code/NonCodingRNA/
Ignored: code/NonCodingRNA_annotation/
Ignored: code/PairwisePi1Traits.P.all.txt.gz
Ignored: code/PeakCalling/
Ignored: code/Phenotypes/
Ignored: code/PlotGruberQTLs/
Ignored: code/PlotQTLs/
Ignored: code/ProCapAnalysis/
Ignored: code/QC/
Ignored: code/QTL_SNP_Enrichment/
Ignored: code/QTLs/
Ignored: code/RPKM_tables/
Ignored: code/ReadLengthMapExperiment/
Ignored: code/ReadLengthMapExperimentResults/
Ignored: code/ReadLengthMapExperimentSpliceCounts/
Ignored: code/ReferenceGenome/
Ignored: code/Rplots.pdf
Ignored: code/Session.vim
Ignored: code/SmallMolecule/
Ignored: code/SplicingAnalysis/
Ignored: code/TODO
Ignored: code/Tehranchi/
Ignored: code/alias/
Ignored: code/bigwigs/
Ignored: code/bigwigs_FromNonWASPFilteredReads/
Ignored: code/config/.DS_Store
Ignored: code/config/._.DS_Store
Ignored: code/config/.ipynb_checkpoints/
Ignored: code/config/config.local.yaml
Ignored: code/dag.pdf
Ignored: code/dag.png
Ignored: code/dag.svg
Ignored: code/data/
Ignored: code/debug.ipynb
Ignored: code/debug_python.ipynb
Ignored: code/deepTools/
Ignored: code/featureCounts/
Ignored: code/featureCountsBasicGtf/
Ignored: code/genome_config.yaml
Ignored: code/gwas_summary_stats/
Ignored: code/hyprcoloc/
Ignored: code/igv_session.xml
Ignored: code/isoseqbams/
Ignored: code/log
Ignored: code/logs/
Ignored: code/notebooks/.ipynb_checkpoints/
Ignored: code/pi1/
Ignored: code/polyA.Splicing.Subset_YRI.NominalPassForColoc.bed.bgz
Ignored: code/rules/.ipynb_checkpoints/
Ignored: code/rules/OldRules/
Ignored: code/rules/notebooks/
Ignored: code/salmontest/
Ignored: code/scratch/
Ignored: code/scripts/.ipynb_checkpoints/
Ignored: code/scripts/GTFtools_0.8.0/
Ignored: code/scripts/__pycache__/
Ignored: code/scripts/liftOverBedpe/liftOverBedpe.py
Ignored: code/snakemake.dryrun.log
Ignored: code/snakemake.log
Ignored: code/snakemake.sbatch.log
Ignored: code/snakemake_profiles/slurm/__pycache__/
Ignored: code/test.introns.bed
Ignored: code/test.introns2.bed
Ignored: code/test.log
Ignored: code/tracks.xml
Ignored: data/.DS_Store
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-10.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-11.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-2.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-3.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-4.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-5.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-6.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-7.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-8.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022.csv
Ignored: data/Metaplots/.DS_Store
Ignored: output/~$20240223_NumQTLs_edited.xlsx
Untracked files:
Untracked: analysis/2024-04-26_eQTL_subtypePlieotropy.Rmd
Untracked: analysis/2024-04-30_MakeExampleConfigForPlotting.Rmd
Untracked: analysis/2024-05-16_IntronSizes.Rmd
Untracked: analysis/2024-06-06_Recreate_naRNA_v_steadyState_juncScatter.Rmd
Untracked: analysis/20240520_ScratchForEditorRequestedFigChanges.Rmd
Untracked: code/scripts/Tidy_GTEx_SummaryStats_AllCisWindowGenes.py
Unstaged changes:
Modified: analysis/2024-01-17_CheckYangsJunctionAnnotator.Rmd
Modified: analysis/2024-04-18_RelativeNMDEfficiency.Rmd
Modified: analysis/MakeFinalFigs_Fig3.Rmd
Modified: analysis/MakeFinalFigs_Fig4.Rmd
Modified: code/rules/GTEx.smk
Modified: code/scripts/GenometracksByGenotype
Modified: code/scripts/SmallMolecule_fitDoseResponseSplicing_AsCassetteExons.R
Modified: code/scripts/Tidy_GTEx_SummaryStats.py
Modified: data/FigS23_Join_Fig5DE.dat.tsv.gz
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/2024-01-17_CheckYangsJunctionAnnotator.Rmd) and
HTML (docs/2024-01-17_CheckYangsJunctionAnnotator.html)
files. If you’ve configured a remote Git repository (see
?wflow_git_remote), click on the hyperlinks in the table
below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 2eb27cd | Benjmain Fair | 2024-04-24 | Update nb after revisions |
| Rmd | 59cbb0e | Benjmain Fair | 2024-03-02 | add tranlsate long reads script |
| Rmd | 39b1eed | Benjmain Fair | 2024-02-13 | add nb for Fig4 |
Intro
Yang wrote a new script to annotate junctions. basically, as I understand it, for each gene he first selects a single functional stop and start codon. Then he considers every set of observed junctions (that are mutually compatible that is) to create every possible transcript and annotates whether each junction creates a PTC in any of those transcripts. I ran the script, using all the junctions oberseved in our sets of experiments (steady state RNA-seq, metabolic labelled, and naRNA-seq), and now I will explore the output.
library(tidyverse)
library(data.table)
library(scattermore)
# Set theme
theme_set(
theme_classic() +
theme(text=element_text(size=16, family="Helvetica")))
# I use layer a lot, to rotate long x-axis labels
Rotate_x_labels <- theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1))
Annotations <- read_tsv("/project/yangili1/bjf79/2024_NMD_Junction_Classifier/Leaf2_junction_classifications.txt") %>%
separate(Intron_coord, into=c("chrom", "start", "end"), sep="[:-]", convert=T, remove = F) %>%
add_count(Intron_coord, name="NumberOfEntriesWithSameCoords")Let’s quickly explore some things about this data frame…
Yang mentioned some introns are duplicated if they overlap multiple genes… He suggested just filtering these out of analysis for simplicity. Let’s count these
Annotations %>%
count(NumberOfEntriesWithSameCoords)# A tibble: 13 × 2
NumberOfEntriesWithSameCoords n
<int> <int>
1 1 297799
2 2 12770
3 3 858
4 4 64
5 5 20
6 6 24
7 7 14
8 8 8
9 9 45
10 15 30
11 17 17
12 20 60
13 22 352The vast majority of introns are listed just once. But as he suggested, let’s filter out the other ones for simplicity.
Annotations.filtered <- Annotations %>%
filter(NumberOfEntriesWithSameCoords==1)
Annotations.filtered %>%
dplyr::select(Intron_coord, Annot, Coding, UTR) %>%
pivot_longer(names_to = "Column", values_to = "BooleanValue", -Intron_coord) %>%
count(Column, BooleanValue) %>%
ggplot(aes(x=Column, y=n, fill=BooleanValue)) +
geom_col(position='fill') +
labs(y="fraction")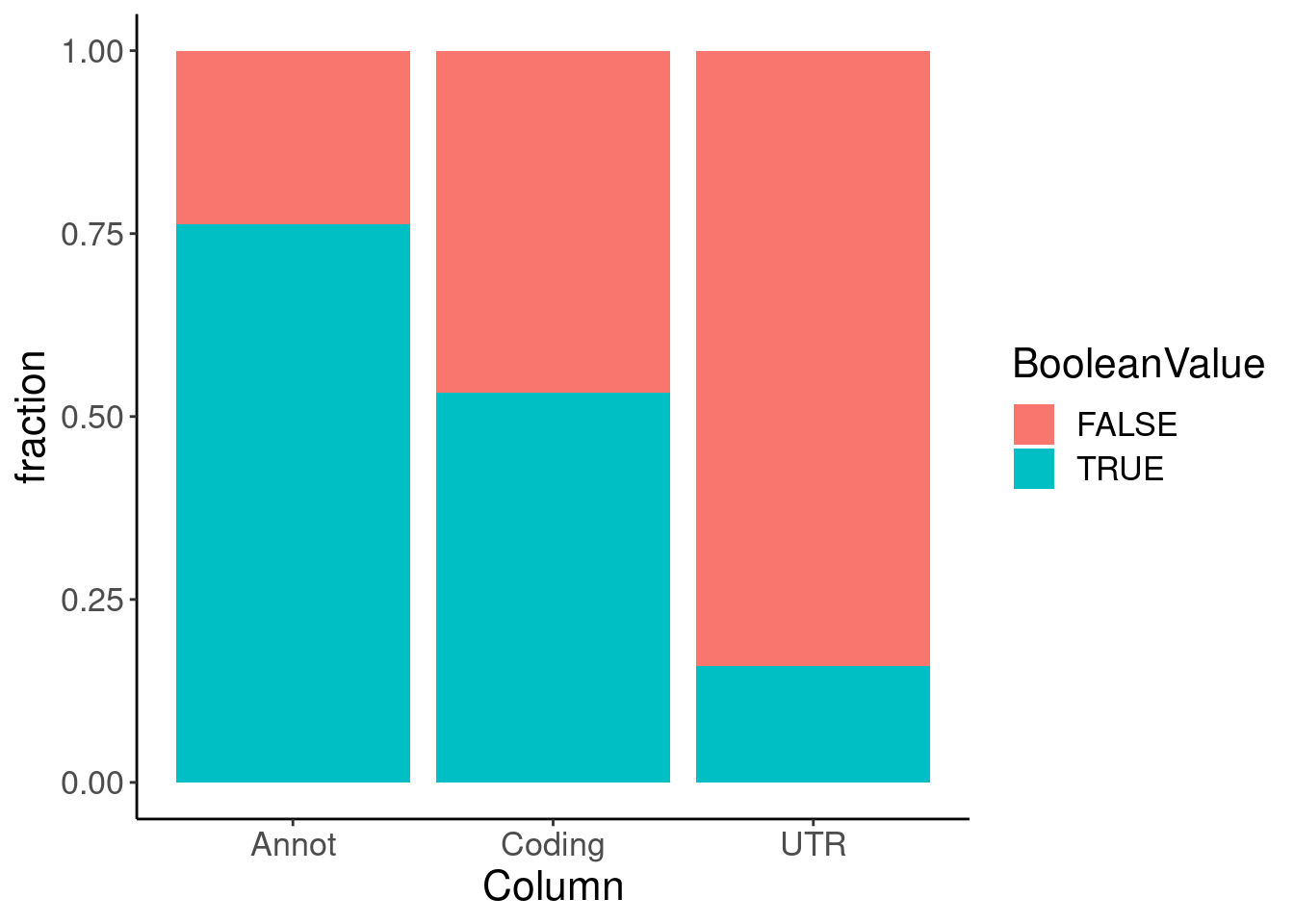
And reminder to self that all UTR junctions (UTR==TRUE) are considered non-coding. Not sure if this is the precise terminology we want in the end… Like UTR junctions are non-coding, but don’t necessarily imply they create a NMD-targeted transcript.
Annotations.filtered %>%
filter(Coding==F) %>%
count(UTR)# A tibble: 2 × 2
UTR n
<lgl> <int>
1 FALSE 91883
2 TRUE 47312Annotations.filtered %>%
filter(Coding) %>%
count(UTR)# A tibble: 1 × 2
UTR n
<lgl> <int>
1 FALSE 158604Annotations.filtered %>%
filter(UTR) %>%
count(Coding)# A tibble: 1 × 2
Coding n
<lgl> <int>
1 FALSE 47312Ok, let’s read in our previous intron annotations and compare…
Previous.Annotations <- read_tsv("../data/IntronAnnotationsFromYang.Updated.tsv.gz")
Annotations.filtered.joined <- Annotations.filtered %>%
inner_join(Previous.Annotations)
Annotations.filtered.joined %>%
distinct(NewAnnotation)# A tibble: 31 × 1
NewAnnotation
<chr>
1 retained_intron.gencode
2 protein_coding.gencode
3 nonsense_mediated_decay.YN
4 processed_transcript.gencode
5 nonsense_mediated_decay.pstopcodon
6 stable.YY
7 nonsense_mediated_decay.far3p
8 stable.UTR_junction
9 nonsense_mediated_decay.far5p
10 nonsense_mediated_decay.gencode
# … with 21 more rowsAnnotations.filtered.joined %>%
distinct(SuperAnnotation)# A tibble: 6 × 1
SuperAnnotation
<chr>
1 AnnotatedJunc_UnproductiveCodingGene
2 AnnotatedJunc_ProductiveCodingGene
3 UnannotatedJunc_UnproductiveCodingGene
4 UnannotatedJunc_ProductiveCodingGene
5 AnnotatedJunc_NoncodingGene
6 UnannotatedJunc_NoncodingJunc Annotations.filtered.joined %>%
count(NewAnnotation, SuperAnnotation, Annot, Coding, UTR) %>%
group_by(NewAnnotation) %>%
mutate(Group_n = sum(n)) %>%
ungroup() %>%
mutate(Color = recode(SuperAnnotation, AnnotatedJunc_NoncodingGene="#6a3d9a", UnannotatedJunc_NoncodingJunc="#cab2d6", AnnotatedJunc_UnproductiveCodingGene="#e31a1c", UnannotatedJunc_UnproductiveCodingGene="#fb9a99", AnnotatedJunc_ProductiveCodingGene="#1f78b4", UnannotatedJunc_ProductiveCodingGene="#a6cee3")) %>%
mutate(groupName = str_glue("n={Group_n}\n{NewAnnotation}")) %>%
filter(Group_n>50) %>%
arrange(SuperAnnotation, Group_n) %>%
pivot_longer(names_to = "Column", values_to = "BooleanValue", c("Annot", "Coding", "UTR")) %>%
ggplot() +
geom_col(aes(x=Column, y=n, fill=BooleanValue), position='fill') +
geom_rect(data = . %>%
distinct(groupName, Color,.keep_all=T), alpha=0,
xmin=-Inf, xmax=Inf, ymin=-Inf, ymax=Inf, size=3, aes(color=Color)) +
scale_color_identity() +
labs(y="fraction") +
facet_wrap(~groupName, scales="free")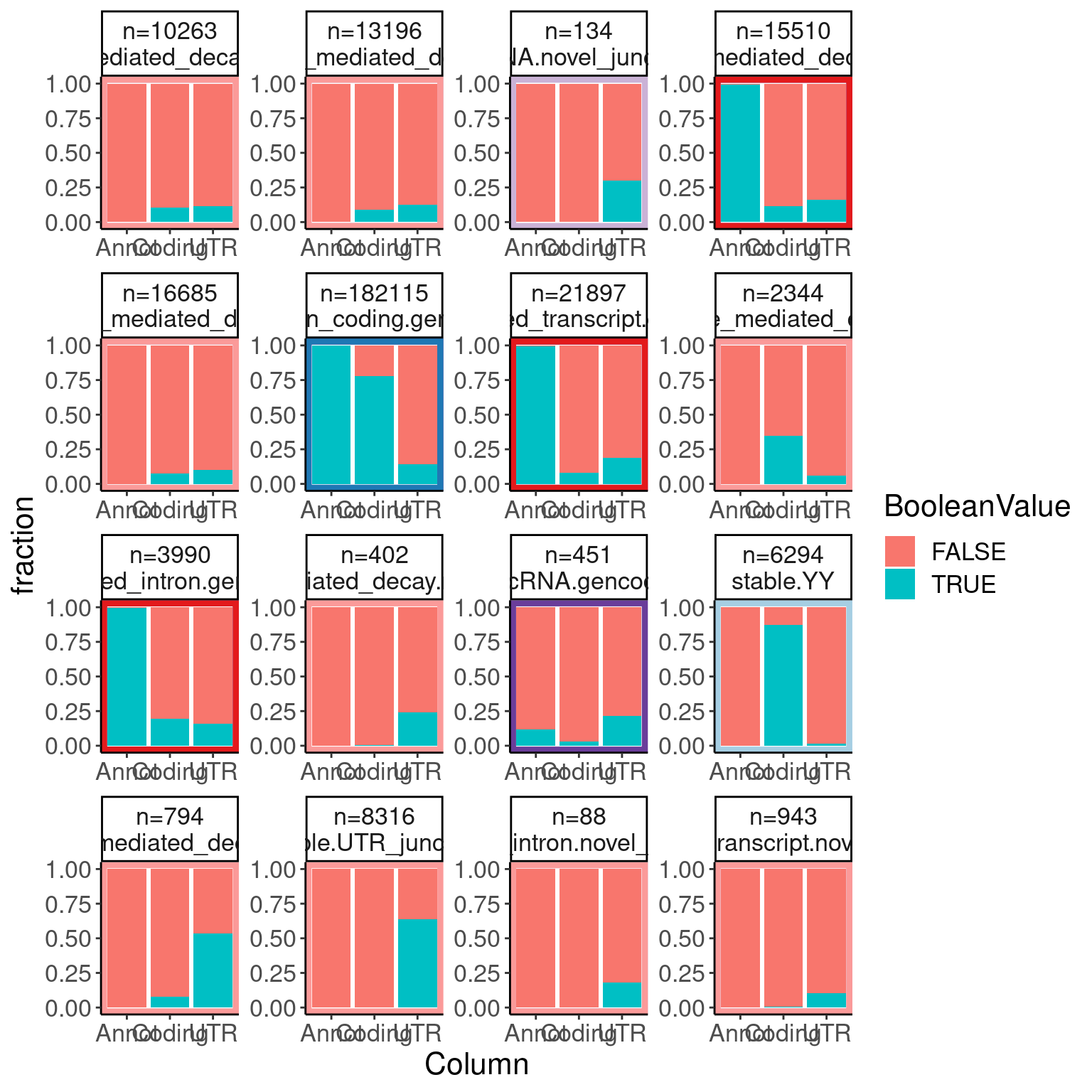
Ok I think those make sense generally. Let’s also look at relative junction usage in naRNA vs steady-state, similar to what we did in a supplement figure with scatter plots of junction RPM.
juncs.long <- fread("../code/SplicingAnalysis/CombinedJuncTables/All.tsv.gz")
dat.KD <- fread("/project2/yangili1/cfbuenabadn/ChromatinSplicingQTLs/code/SplicingAnalysis/CombinedJuncTables/NMD_KD.tsv.gz")
juncs.long.summary <-
bind_rows(
juncs.long,
dat.KD) %>%
dplyr::select(chrom, start, stop, strand, Dataset, Count) %>%
group_by(Dataset, chrom, start, stop) %>%
summarise(Sum=sum(Count)) %>%
ungroup()
juncs.long.summary.joined <- juncs.long.summary %>%
group_by(Dataset) %>%
mutate(DatasetSum = sum(Sum)) %>%
ungroup() %>%
mutate(RPM = Sum/DatasetSum*1E6) %>%
mutate(stop = stop+1) %>%
inner_join(Annotations.filtered.joined, by=c("chrom", "start", "stop"="end"))
juncs.long.summary.joined %>%
distinct(Dataset)# A tibble: 9 × 1
Dataset
<chr>
1 Expression.Splicing
2 HeLa.SMG6.KD
3 HeLa.SMG7.KD
4 HeLa.UPF1.KD
5 HeLa.dKD
6 HeLa.scr
7 MetabolicLabelled.30min
8 MetabolicLabelled.60min
9 chRNA.Expression.Splicingjuncs.long.summary.joined %>%
filter(!UTR) %>%
filter(Dataset %in% c("Expression.Splicing", "chRNA.Expression.Splicing")) %>%
dplyr::select(-DatasetSum, -Sum) %>%
pivot_wider(names_from="Dataset", values_from="RPM") %>%
mutate(Color = recode(SuperAnnotation, AnnotatedJunc_NoncodingGene="#6a3d9a", UnannotatedJunc_NoncodingJunc="#cab2d6", AnnotatedJunc_UnproductiveCodingGene="#e31a1c", UnannotatedJunc_UnproductiveCodingGene="#fb9a99", AnnotatedJunc_ProductiveCodingGene="#1f78b4", UnannotatedJunc_ProductiveCodingGene="#a6cee3")) %>%
mutate(SuperAnnotation = factor(SuperAnnotation, levels=c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene", "AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene", "AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))) %>%
ggplot(aes(x=Expression.Splicing, y=chRNA.Expression.Splicing)) +
geom_point(alpha=0.01, color='black') +
geom_density2d(aes(color=Color)) +
geom_text(data = . %>%
replace_na(list(Expression.Splicing=1E-5, chRNA.Expression.Splicing=1E-5)) %>%
group_by(Coding, SuperAnnotation) %>%
summarise(med = round(median(chRNA.Expression.Splicing/Expression.Splicing, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("med={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
geom_abline(slope = 1, color='red') +
facet_grid(Coding~SuperAnnotation) +
theme_bw() +
labs(x="Junction RPM, steady-state", y="Junction RPM, chRNA", caption="Filtered out UTR juncs.\nHorizontal facets are 'Coding' value")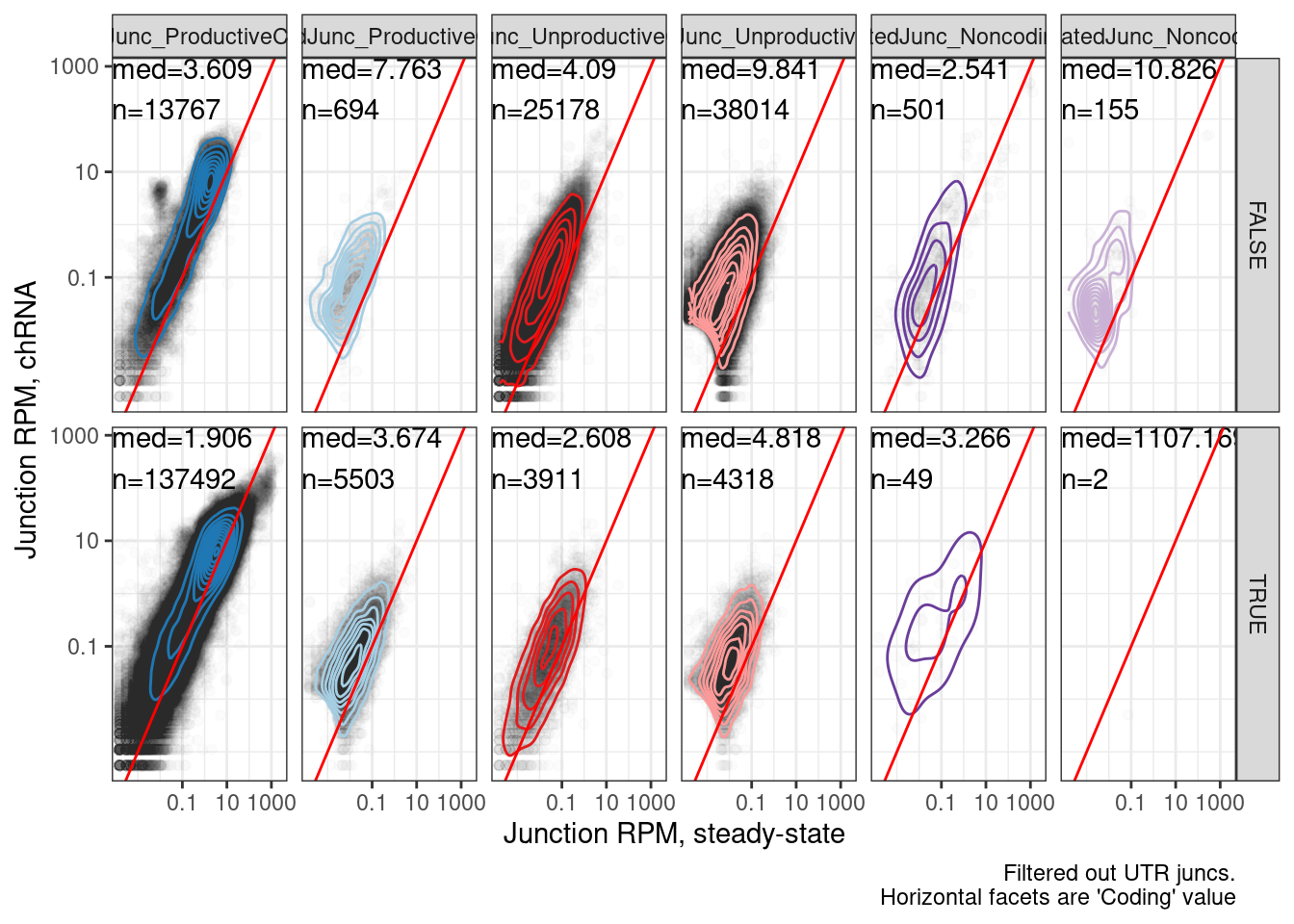
juncs.long.summary.joined %>%
filter(!UTR) %>%
filter(Dataset %in% c("HeLa.scr", "HeLa.dKD")) %>%
dplyr::select(-DatasetSum, -Sum) %>%
pivot_wider(names_from="Dataset", values_from="RPM") %>%
mutate(Color = recode(SuperAnnotation, AnnotatedJunc_NoncodingGene="#6a3d9a", UnannotatedJunc_NoncodingJunc="#cab2d6", AnnotatedJunc_UnproductiveCodingGene="#e31a1c", UnannotatedJunc_UnproductiveCodingGene="#fb9a99", AnnotatedJunc_ProductiveCodingGene="#1f78b4", UnannotatedJunc_ProductiveCodingGene="#a6cee3")) %>%
mutate(SuperAnnotation = factor(SuperAnnotation, levels=c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene", "AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene", "AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))) %>%
ggplot(aes(x=HeLa.scr, y=HeLa.dKD)) +
geom_point(alpha=0.01, color='black') +
geom_density2d(aes(color=Color)) +
geom_text(data = . %>%
replace_na(list(HeLa.scr=1E-5, HeLa.dKD=1E-5)) %>%
group_by(Coding, SuperAnnotation) %>%
summarise(med = round(median(HeLa.dKD/HeLa.scr, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("med={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
geom_abline(slope = 1, color='red') +
facet_grid(Coding~SuperAnnotation) +
theme_bw() +
labs(x="Junction RPM, control", y="Junction RPM, dKD", caption="Filtered out UTR juncs.\nHorizontal facets are 'Coding' value")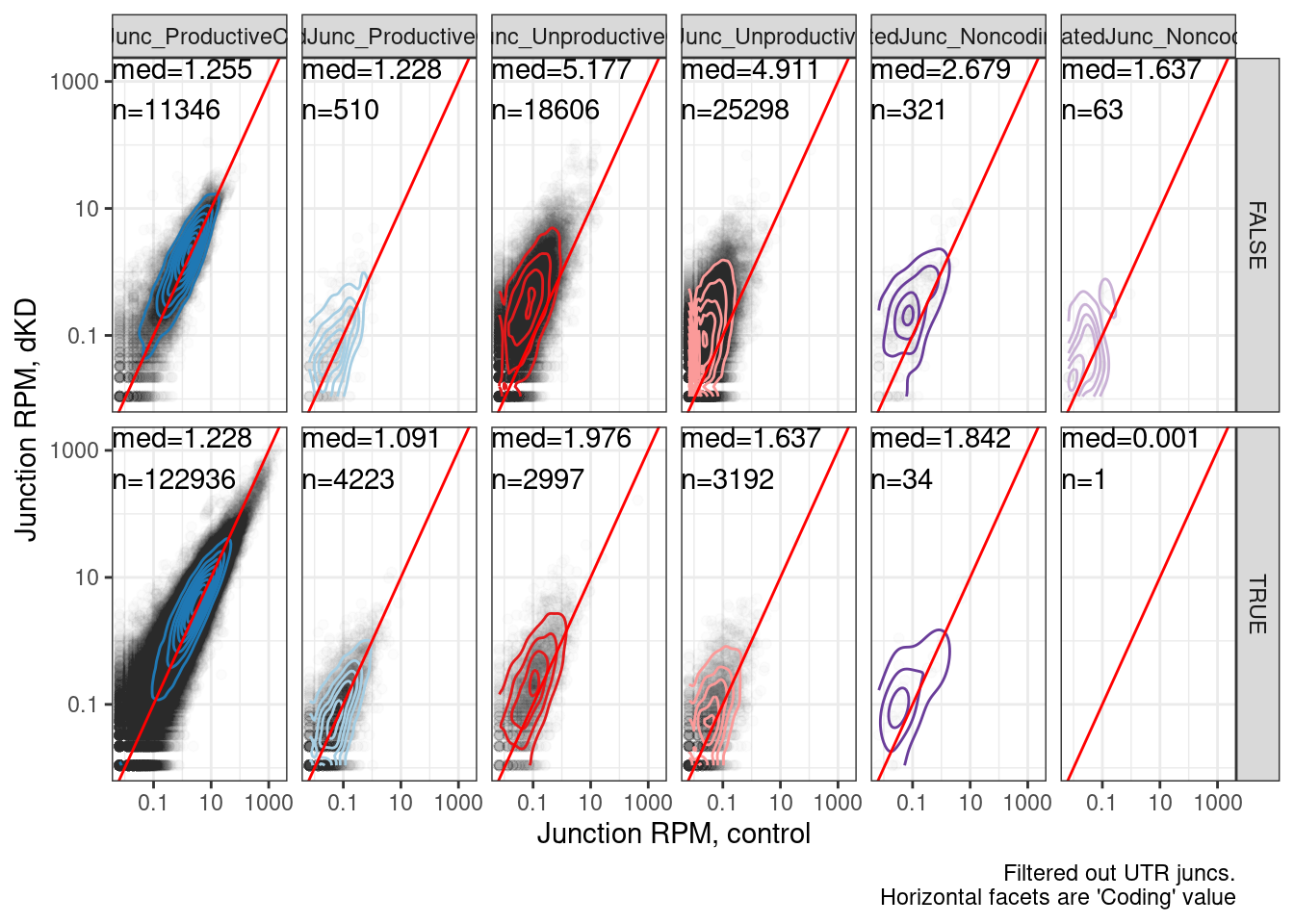
Response to review
Let’s make similar versions of these plots but now from the long read-determined context of juncs
JuncAnnotationsFromLongReadContext <- read_tsv("../output/20240322_ResponseToReviewerMostCommonJuncContexts.tsv.gz") %>%
separate(Introns, into=c("chrom", "start", "stop", "strand"), sep="_", convert=T)
juncs.long.summary.joined.LongReadContext <- juncs.long.summary %>%
group_by(Dataset) %>%
mutate(DatasetSum = sum(Sum)) %>%
ungroup() %>%
mutate(RPM = Sum/DatasetSum*1E6) %>%
mutate(stop = stop+1) %>%
inner_join(JuncAnnotationsFromLongReadContext, by=c("chrom", "start", "stop"))
juncs.long.summary.joined.LongReadContext %>%
distinct(Dataset)# A tibble: 9 × 1
Dataset
<chr>
1 Expression.Splicing
2 HeLa.SMG6.KD
3 HeLa.SMG7.KD
4 HeLa.UPF1.KD
5 HeLa.dKD
6 HeLa.scr
7 MetabolicLabelled.30min
8 MetabolicLabelled.60min
9 chRNA.Expression.Splicingjuncs.long.summary.joined.LongReadContext %>%
filter(Dataset %in% c("Expression.Splicing", "chRNA.Expression.Splicing")) %>%
dplyr::select(-DatasetSum, -Sum) %>%
pivot_wider(names_from="Dataset", values_from="RPM") %>%
mutate(Color = recode(SuperAnnotation, AnnotatedJunc_NoncodingGene="#6a3d9a", UnannotatedJunc_NoncodingJunc="#cab2d6", AnnotatedJunc_UnproductiveCodingGene="#e31a1c", UnannotatedJunc_UnproductiveCodingGene="#fb9a99", AnnotatedJunc_ProductiveCodingGene="#1f78b4", UnannotatedJunc_ProductiveCodingGene="#a6cee3")) %>%
mutate(SuperAnnotation = factor(SuperAnnotation, levels=c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene", "AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene", "AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))) %>%
ggplot(aes(x=Expression.Splicing, y=chRNA.Expression.Splicing)) +
geom_point(alpha=0.01, color='black') +
geom_density2d() +
geom_text(data = . %>%
replace_na(list(Expression.Splicing=1E-5, chRNA.Expression.Splicing=1E-5)) %>%
group_by(ModeNMDFinder, SuperAnnotation) %>%
summarise(med = round(median(chRNA.Expression.Splicing/Expression.Splicing, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("med={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
geom_abline(slope = 1, color='red') +
facet_grid(ModeNMDFinder~SuperAnnotation) +
theme_bw() +
labs(x="Junction RPM, steady-state", y="Junction RPM, chRNA")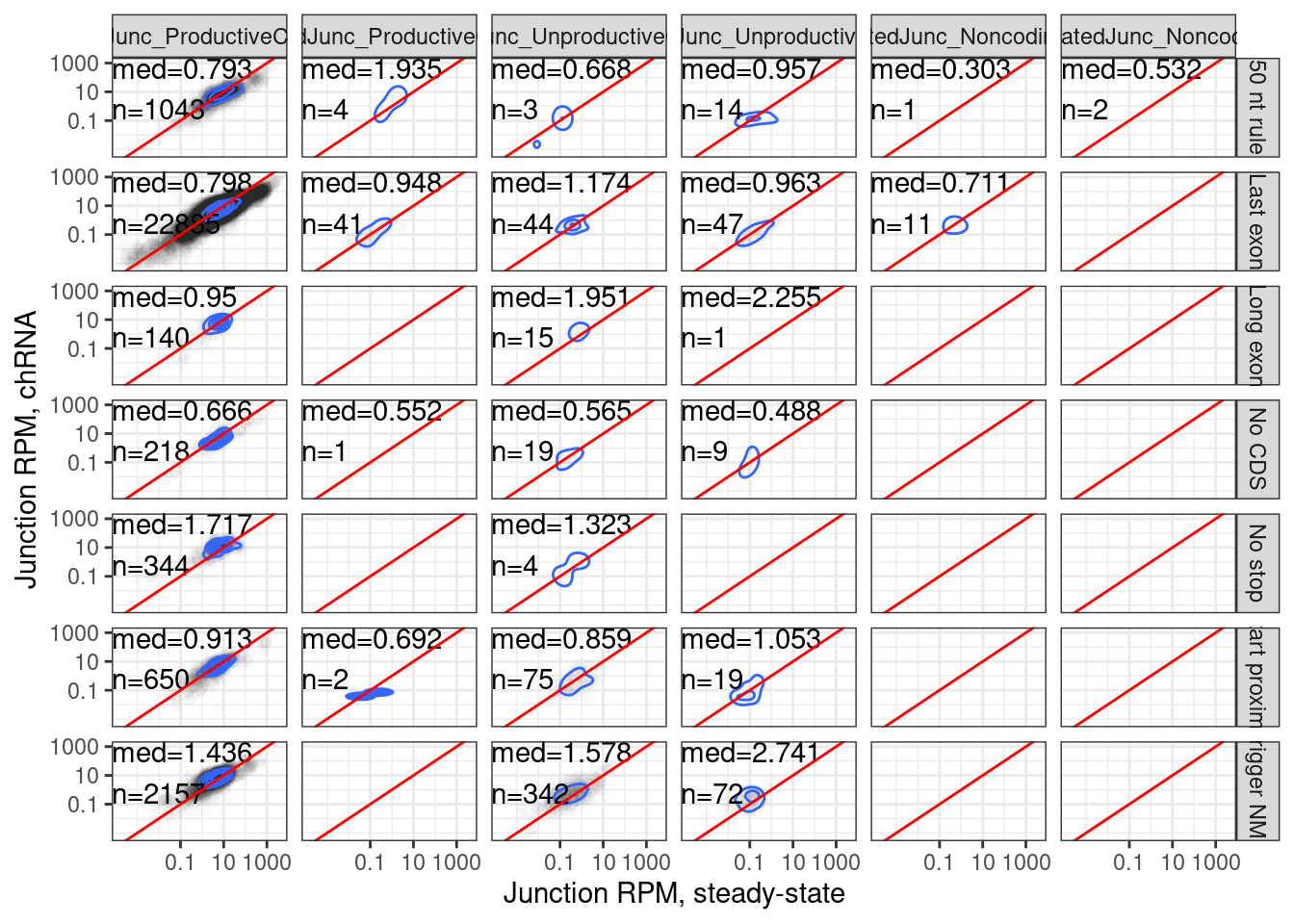
juncs.long.summary.joined.LongReadContext %>%
filter(Dataset %in% c("HeLa.scr", "HeLa.dKD")) %>%
dplyr::select(-DatasetSum, -Sum) %>%
pivot_wider(names_from="Dataset", values_from="RPM") %>%
mutate(Color = recode(SuperAnnotation, AnnotatedJunc_NoncodingGene="#6a3d9a", UnannotatedJunc_NoncodingJunc="#cab2d6", AnnotatedJunc_UnproductiveCodingGene="#e31a1c", UnannotatedJunc_UnproductiveCodingGene="#fb9a99", AnnotatedJunc_ProductiveCodingGene="#1f78b4", UnannotatedJunc_ProductiveCodingGene="#a6cee3")) %>%
mutate(SuperAnnotation = factor(SuperAnnotation, levels=c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene", "AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene", "AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))) %>%
ggplot(aes(x=HeLa.scr, y=HeLa.dKD)) +
geom_point(alpha=0.01, color='black') +
geom_density2d() +
geom_text(data = . %>%
replace_na(list(HeLa.scr=1E-5, HeLa.dKD=1E-5)) %>%
group_by(ModeNMDFinder, SuperAnnotation) %>%
summarise(med = round(median(HeLa.dKD/HeLa.scr, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("med={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
geom_abline(slope = 1, color='red') +
facet_grid(ModeNMDFinder~SuperAnnotation) +
theme_bw() +
labs(x="Junction RPM, control", y="Junction RPM, dKD")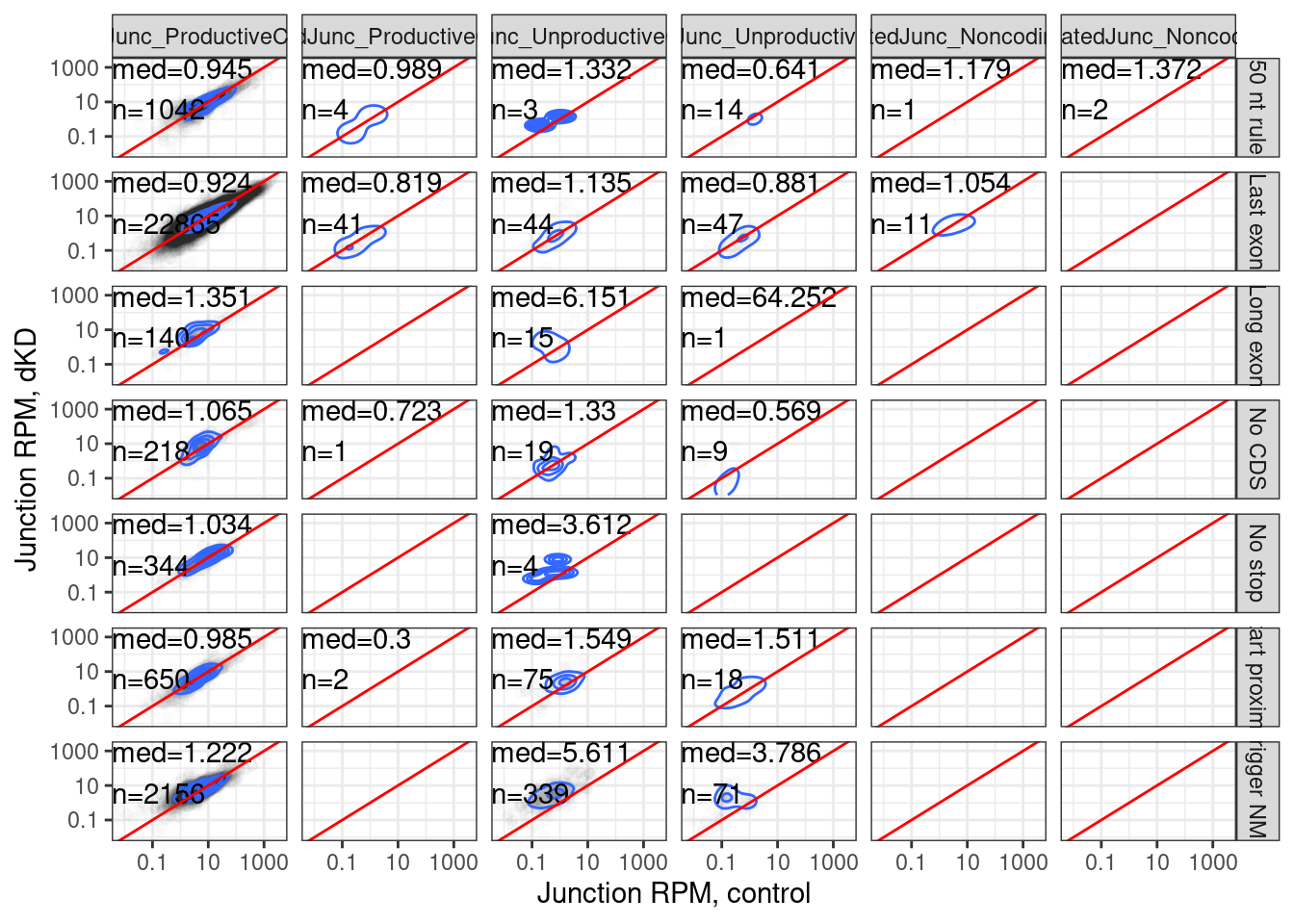
…same thing but now just disregard the old categories
juncs.long.summary.joined.LongReadContext %>%
filter(Dataset %in% c("Expression.Splicing", "chRNA.Expression.Splicing")) %>%
dplyr::select(-DatasetSum, -Sum) %>%
pivot_wider(names_from="Dataset", values_from="RPM") %>%
ggplot(aes(x=Expression.Splicing, y=chRNA.Expression.Splicing)) +
geom_point(alpha=0.01, color='black') +
geom_density2d() +
geom_text(data = . %>%
replace_na(list(Expression.Splicing=1E-5, chRNA.Expression.Splicing=1E-5)) %>%
group_by(ModeNMDFinder) %>%
summarise(med = round(median(chRNA.Expression.Splicing/Expression.Splicing, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("med={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
geom_abline(slope = 1, color='red') +
facet_wrap(~ModeNMDFinder) +
theme_bw() +
labs(x="Junction RPM, steady-state", y="Junction RPM, chRNA")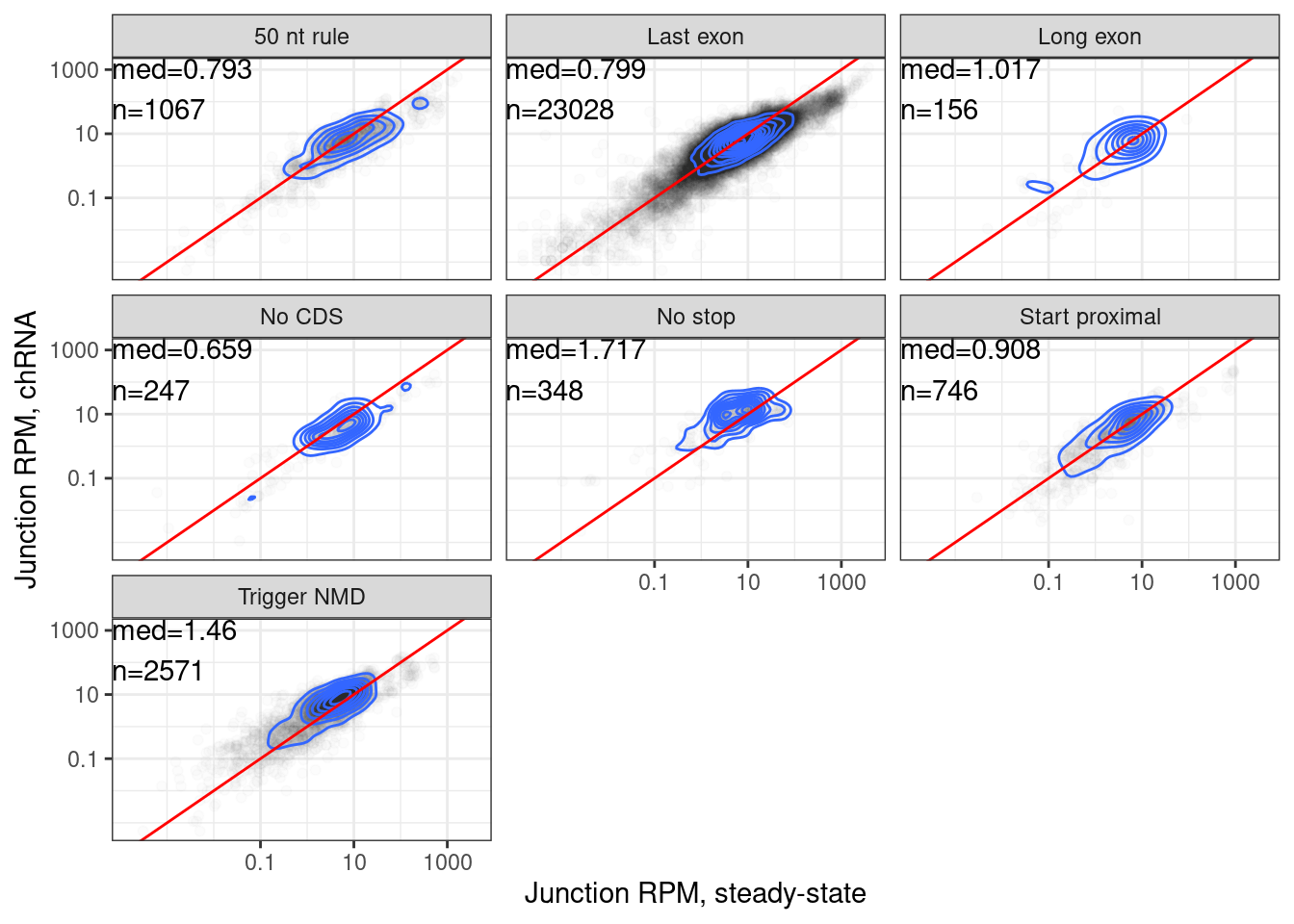
juncs.long.summary.joined.LongReadContext %>%
filter(Dataset %in% c("HeLa.scr", "HeLa.dKD")) %>%
dplyr::select(-DatasetSum, -Sum) %>%
pivot_wider(names_from="Dataset", values_from="RPM") %>%
ggplot(aes(x=HeLa.scr, y=HeLa.dKD)) +
geom_point(alpha=0.01, color='black') +
geom_density2d() +
geom_text(data = . %>%
replace_na(list(HeLa.scr=1E-5, HeLa.dKD=1E-5)) %>%
group_by(ModeNMDFinder) %>%
summarise(med = round(median(HeLa.dKD/HeLa.scr, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("med={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
geom_abline(slope = 1, color='red') +
facet_wrap(~ModeNMDFinder) +
theme_bw() +
labs(x="Junction RPM, control", y="Junction RPM, dKD")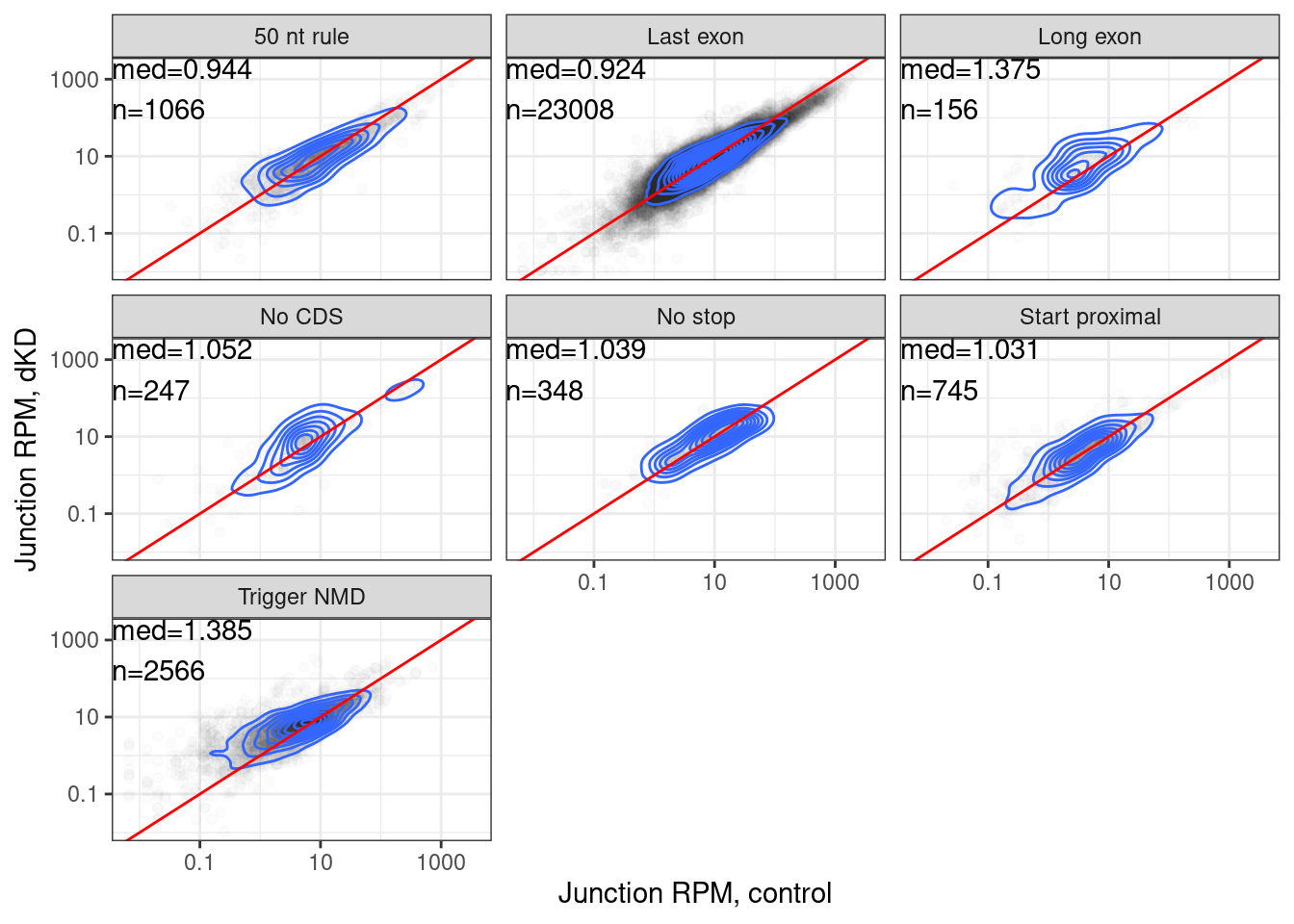 Ok let’s plot these median fold enrichments as a heatmap, since it is
totally consistent with the levels of NMD efficiency observed in
Lindeboom et al.
Ok let’s plot these median fold enrichments as a heatmap, since it is
totally consistent with the levels of NMD efficiency observed in
Lindeboom et al.
NMD.Efficiency.P.dat <- bind_rows(
juncs.long.summary.joined.LongReadContext %>%
filter(Dataset %in% c("Expression.Splicing", "chRNA.Expression.Splicing")) %>%
dplyr::select(-DatasetSum, -Sum) %>%
pivot_wider(names_from="Dataset", values_from="RPM") %>%
replace_na(list(Expression.Splicing=1E-5, chRNA.Expression.Splicing=1E-5)) %>%
group_by(ModeNMDFinder) %>%
summarise(med = log2(median(chRNA.Expression.Splicing/Expression.Splicing, na.rm=F))) %>%
ungroup() %>%
mutate(Comparison = "log2(naRNA/SteadyState)"),
juncs.long.summary.joined.LongReadContext %>%
filter(Dataset %in% c("HeLa.scr", "HeLa.dKD")) %>%
dplyr::select(-DatasetSum, -Sum) %>%
pivot_wider(names_from="Dataset", values_from="RPM") %>%
replace_na(list(HeLa.scr=1E-5, HeLa.dKD=1E-5)) %>%
group_by(ModeNMDFinder) %>%
summarise(med = log2(median(HeLa.dKD/HeLa.scr, na.rm=F))) %>%
ungroup() %>%
mutate(Comparison = "log2(HeLa.dKD/HeLa.scr)")
)
NMD.Efficiency.P.dat %>%
inner_join(NMD.Efficiency.P.dat %>%
filter(ModeNMDFinder == "Last exon") %>%
dplyr::select(LastExonMed = med, Comparison)) %>%
mutate(MedEffectRelativeToLastExonMed = med - LastExonMed) %>%
mutate(ModeNMDFinder = factor(ModeNMDFinder, levels=c("Last exon", "50 nt rule", "Start proximal", "Long exon", "Trigger NMD", "No stop", "No CDS"))) %>%
mutate(Comparison = recode(Comparison, "log2(HeLa.dKD/HeLa.scr)"=str_wrap("shRNA control\nvs\nshRNA dKD NMD factors", 10), "log2(naRNA/SteadyState)"=str_wrap("steady-state vs naRNA", 20))) %>%
ggplot(aes(x=Comparison, y=ModeNMDFinder, fill=MedEffectRelativeToLastExonMed)) +
geom_raster() +
geom_text(aes(label=round(MedEffectRelativeToLastExonMed, 3))) +
scale_fill_viridis_c() +
scale_x_discrete(expand=c(0,0)) +
scale_y_discrete(expand=c(0,0)) +
labs(y="Most common full-\ntranscript context", x="Short read datasets for degredation efficiency estimate", fill="Degredation\nefficiency\nestimate")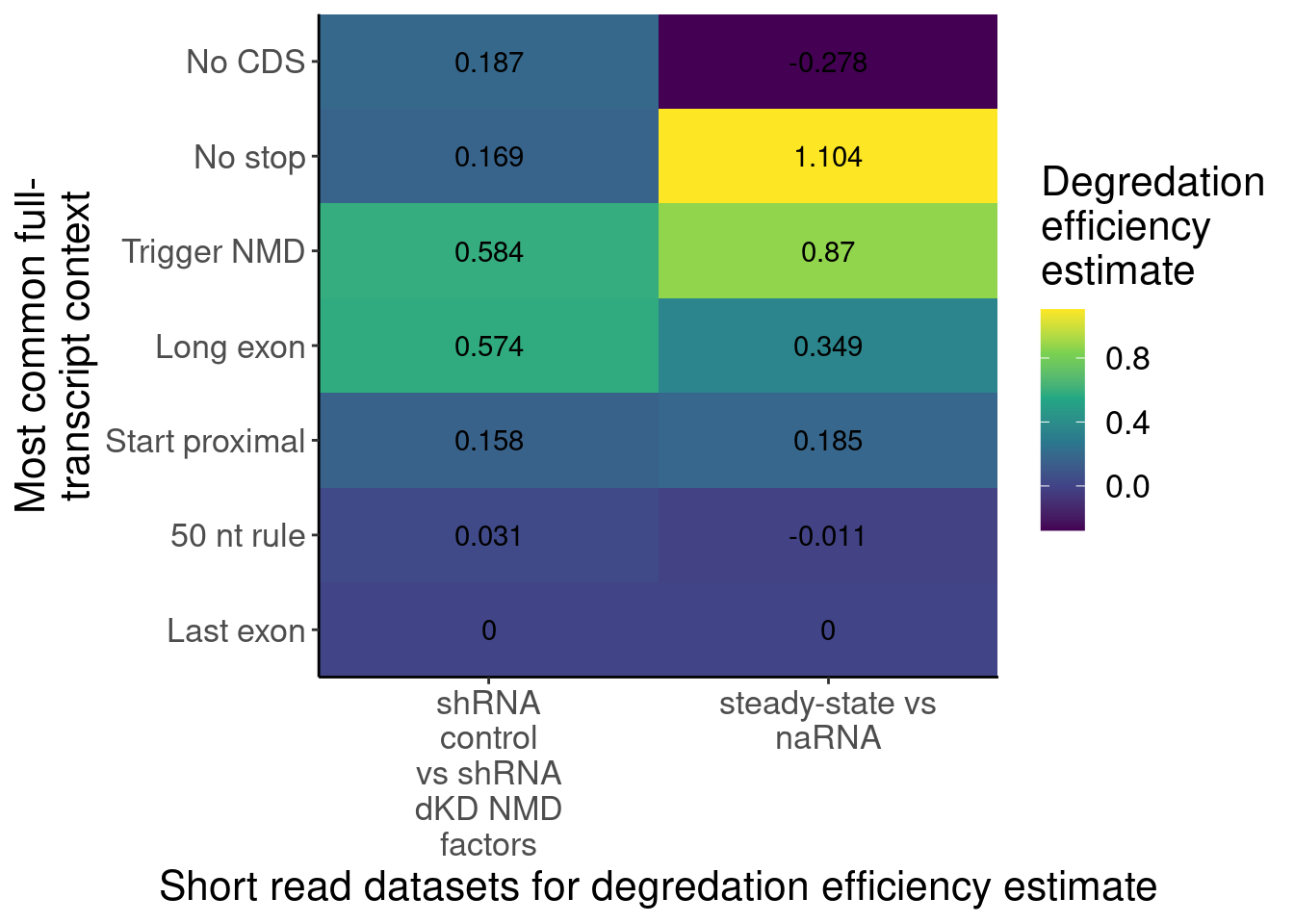
ggsave("/project2/yangili1/carlos_and_ben_shared/rough_figs/OriginalSubplots/202403_ResponseToReviewersDegredationEfficiency.pdf", width=5, height=4)Head to head plots
Let’s more directly plot the old annotations vs the new annotations to see which method is better (has more enrichment in dKD or chRNA)
juncs.long.summary.joined.toplot <- juncs.long.summary.joined %>%
filter(!UTR) %>%
filter(Dataset %in% c("HeLa.scr", "HeLa.dKD")) %>%
dplyr::select(-DatasetSum, -Sum) %>%
pivot_wider(names_from="Dataset", values_from="RPM") %>%
mutate(Color = recode(SuperAnnotation, AnnotatedJunc_NoncodingGene="#6a3d9a", UnannotatedJunc_NoncodingJunc="#cab2d6", AnnotatedJunc_UnproductiveCodingGene="#e31a1c", UnannotatedJunc_UnproductiveCodingGene="#fb9a99", AnnotatedJunc_ProductiveCodingGene="#1f78b4", UnannotatedJunc_ProductiveCodingGene="#a6cee3")) %>%
mutate(SuperAnnotation = factor(SuperAnnotation, levels=c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene", "AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene", "AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))) %>%
filter(!SuperAnnotation %in% c("AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))
bind_rows(
juncs.long.summary.joined.toplot %>%
mutate(method="Old"),
juncs.long.summary.joined.toplot %>%
mutate(method="New"),
) %>%
mutate(ProductiveOrUnproductive = case_when(
method == "New" ~ Coding,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene") ~ TRUE,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene") ~ FALSE
)) %>%
ggplot(aes(x=HeLa.scr, y=HeLa.dKD)) +
geom_point(alpha=0.01, color='black') +
geom_density2d(color='red') +
geom_text(data = . %>%
replace_na(list(HeLa.scr=1E-5, HeLa.dKD=1E-5)) %>%
group_by(method, ProductiveOrUnproductive) %>%
summarise(med = round(median(HeLa.dKD/HeLa.scr, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("med={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
geom_abline(slope = 1, color='red') +
facet_grid(method~ProductiveOrUnproductive) +
theme_bw() +
labs(x="Junction RPM, control", y="Junction RPM, dKD", caption="Filtered out UTR juncs.\nVertical facets are 'Coding' value")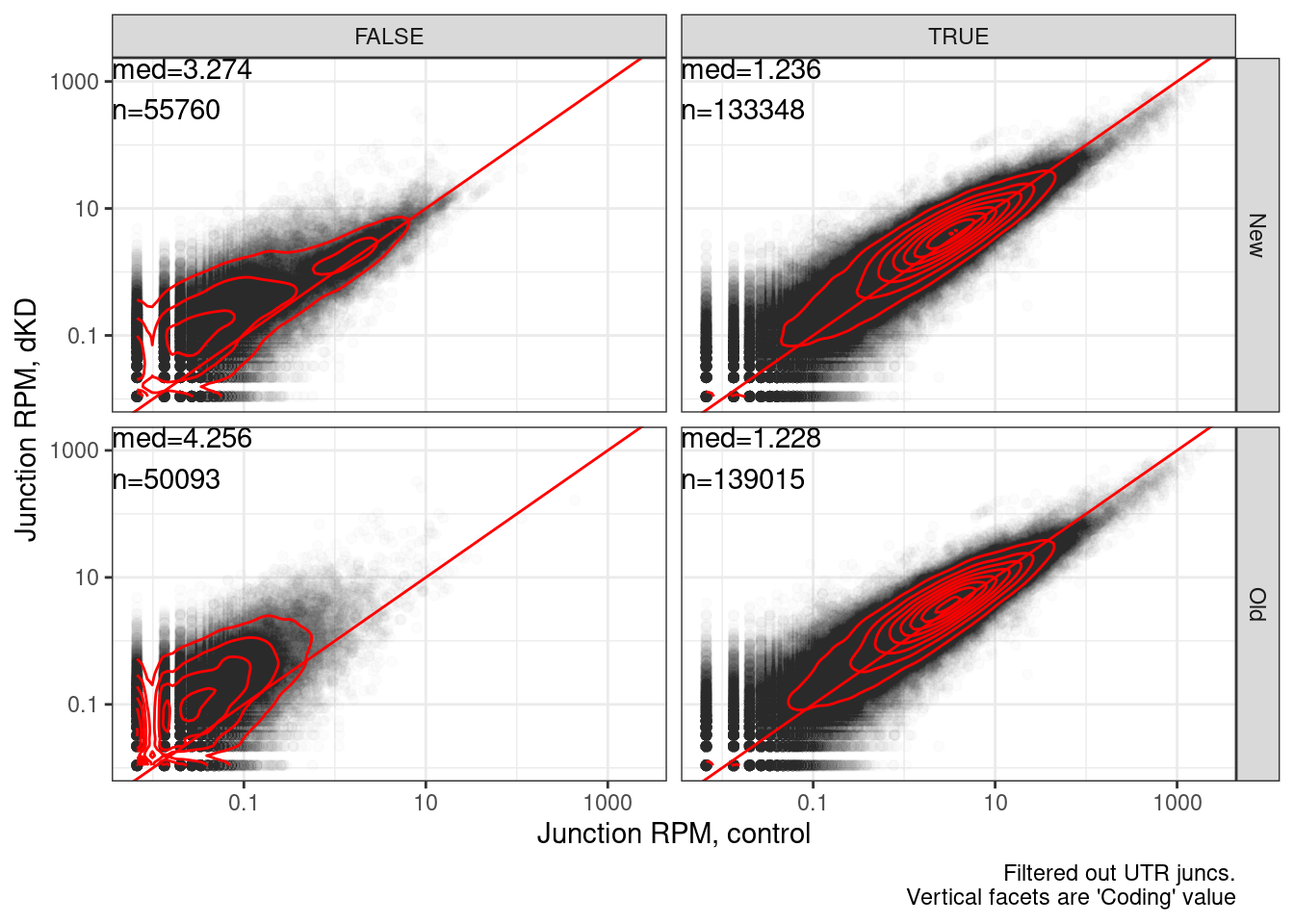
Same plot but steady state vs chRNA…
juncs.long.summary.joined.toplot <- juncs.long.summary.joined %>%
filter(!UTR) %>%
filter(Dataset %in% c("Expression.Splicing", "chRNA.Expression.Splicing")) %>%
dplyr::select(-DatasetSum, -Sum) %>%
pivot_wider(names_from="Dataset", values_from="RPM") %>%
mutate(Color = recode(SuperAnnotation, AnnotatedJunc_NoncodingGene="#6a3d9a", UnannotatedJunc_NoncodingJunc="#cab2d6", AnnotatedJunc_UnproductiveCodingGene="#e31a1c", UnannotatedJunc_UnproductiveCodingGene="#fb9a99", AnnotatedJunc_ProductiveCodingGene="#1f78b4", UnannotatedJunc_ProductiveCodingGene="#a6cee3")) %>%
mutate(SuperAnnotation = factor(SuperAnnotation, levels=c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene", "AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene", "AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))) %>%
filter(!SuperAnnotation %in% c("AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))
bind_rows(
juncs.long.summary.joined.toplot %>%
mutate(method="Old"),
juncs.long.summary.joined.toplot %>%
mutate(method="New"),
) %>%
mutate(ProductiveOrUnproductive = case_when(
method == "New" ~ Coding,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene") ~ TRUE,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene") ~ FALSE
)) %>%
ggplot(aes(x=Expression.Splicing, y=chRNA.Expression.Splicing)) +
geom_point(alpha=0.01, color='black') +
geom_density2d(color='red') +
geom_text(data = . %>%
replace_na(list(Expression.Splicing=1E-5, chRNA.Expression.Splicing=1E-5)) %>%
group_by(method, ProductiveOrUnproductive) %>%
summarise(med = round(median(chRNA.Expression.Splicing/Expression.Splicing, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("med={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
geom_abline(slope = 1, color='red') +
facet_grid(method~ProductiveOrUnproductive) +
theme_bw() +
labs(x="Junction RPM, steady-state", y="Junction RPM, chRNA", caption="Filtered out UTR juncs.\nVertical facets are 'Coding' value")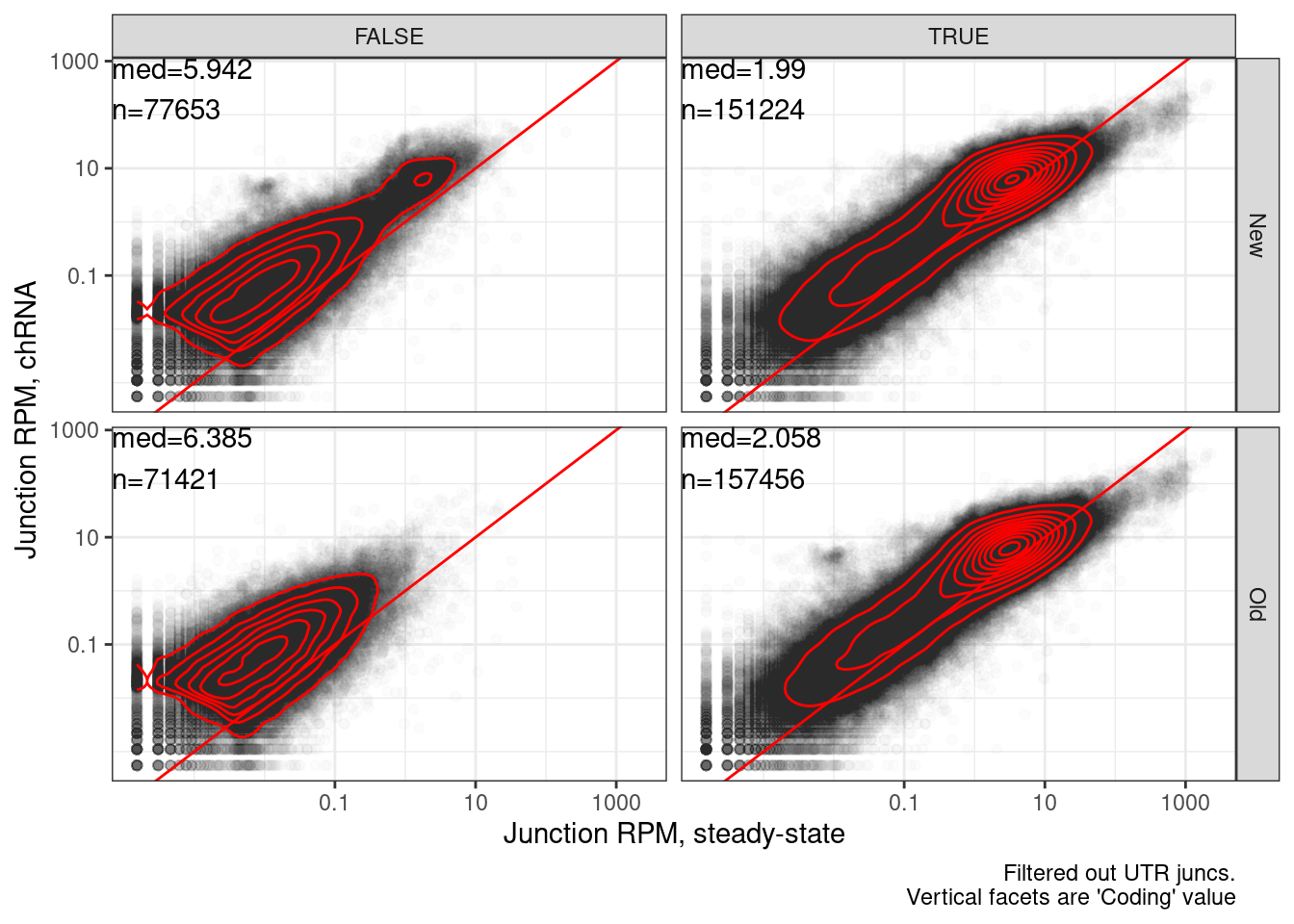
Head to head, with more inclusive for new coding method
Yang suggested tweaking the New classification to also include all annotated productive juncs and productive. So an or statement… here is pseudocode: IsProductiveInNewMethod = (Coding==True OR AnnotatedProductive==TRUE). Now let’s remake those head to head plots…
juncs.long.summary.joined.toplot <- juncs.long.summary.joined %>%
filter(!UTR) %>%
filter(Dataset %in% c("HeLa.scr", "HeLa.dKD")) %>%
dplyr::select(-DatasetSum, -Sum) %>%
pivot_wider(names_from="Dataset", values_from="RPM") %>%
mutate(Color = recode(SuperAnnotation, AnnotatedJunc_NoncodingGene="#6a3d9a", UnannotatedJunc_NoncodingJunc="#cab2d6", AnnotatedJunc_UnproductiveCodingGene="#e31a1c", UnannotatedJunc_UnproductiveCodingGene="#fb9a99", AnnotatedJunc_ProductiveCodingGene="#1f78b4", UnannotatedJunc_ProductiveCodingGene="#a6cee3")) %>%
mutate(SuperAnnotation = factor(SuperAnnotation, levels=c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene", "AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene", "AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))) %>%
filter(!SuperAnnotation %in% c("AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))
bind_rows(
juncs.long.summary.joined.toplot %>%
mutate(method="Old"),
juncs.long.summary.joined.toplot %>%
mutate(method="New"),
) %>%
mutate(ProductiveOrUnproductive = case_when(
method == "New" & (Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene") ~ TRUE,
method == "New" & !(Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene") ~ FALSE,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene") ~ TRUE,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene") ~ FALSE
)) %>%
ggplot(aes(x=HeLa.scr, y=HeLa.dKD)) +
geom_point(alpha=0.01, color='black') +
geom_density2d(color='red') +
geom_text(data = . %>%
replace_na(list(HeLa.scr=1E-5, HeLa.dKD=1E-5)) %>%
group_by(method, ProductiveOrUnproductive) %>%
summarise(med = round(median(HeLa.dKD/HeLa.scr, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("med={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
geom_abline(slope = 1, color='red') +
facet_grid(method~ProductiveOrUnproductive) +
theme_bw() +
labs(x="Junction RPM, control", y="Junction RPM, dKD", caption="Filtered out UTR juncs.\nVertical facets are 'Coding' value")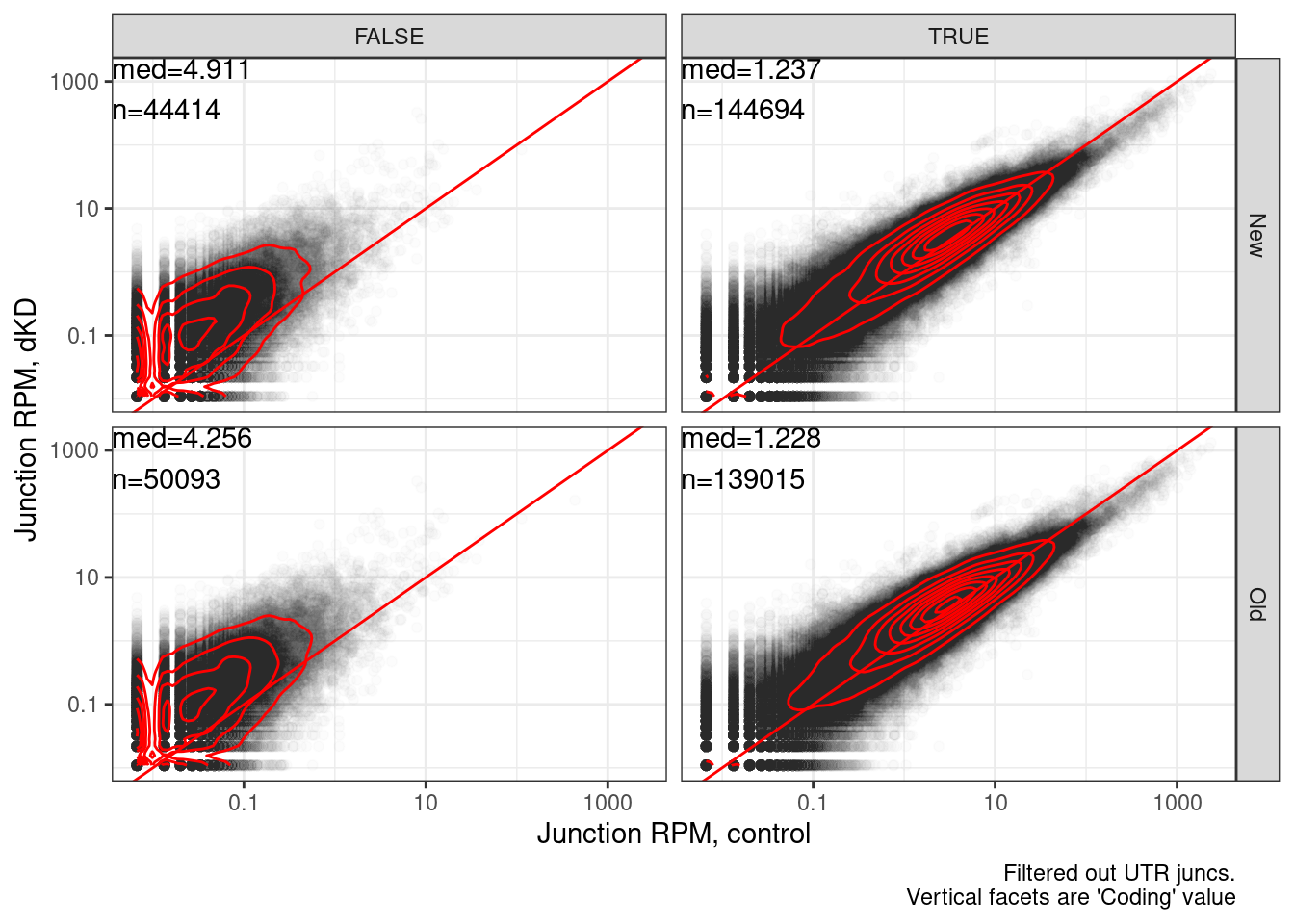
Same plot but steady state vs chRNA…
juncs.long.summary.joined.toplot <- juncs.long.summary.joined %>%
filter(!UTR) %>%
filter(Dataset %in% c("Expression.Splicing", "chRNA.Expression.Splicing")) %>%
dplyr::select(-DatasetSum, -Sum) %>%
pivot_wider(names_from="Dataset", values_from="RPM") %>%
mutate(Color = recode(SuperAnnotation, AnnotatedJunc_NoncodingGene="#6a3d9a", UnannotatedJunc_NoncodingJunc="#cab2d6", AnnotatedJunc_UnproductiveCodingGene="#e31a1c", UnannotatedJunc_UnproductiveCodingGene="#fb9a99", AnnotatedJunc_ProductiveCodingGene="#1f78b4", UnannotatedJunc_ProductiveCodingGene="#a6cee3")) %>%
mutate(SuperAnnotation = factor(SuperAnnotation, levels=c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene", "AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene", "AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))) %>%
filter(!SuperAnnotation %in% c("AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))
bind_rows(
juncs.long.summary.joined.toplot %>%
mutate(method="Old"),
juncs.long.summary.joined.toplot %>%
mutate(method="New"),
) %>%
mutate(ProductiveOrUnproductive = case_when(
method == "New" & (Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene") ~ TRUE,
method == "New" & !(Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene") ~ FALSE,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene") ~ TRUE,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene") ~ FALSE
)) %>%
ggplot(aes(x=Expression.Splicing, y=chRNA.Expression.Splicing)) +
geom_point(alpha=0.01, color='black') +
geom_density2d(color='red') +
geom_text(data = . %>%
replace_na(list(Expression.Splicing=1E-5, chRNA.Expression.Splicing=1E-5)) %>%
group_by(method, ProductiveOrUnproductive) %>%
summarise(med = round(median(chRNA.Expression.Splicing/Expression.Splicing, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("med={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
geom_abline(slope = 1, color='red') +
facet_grid(method~ProductiveOrUnproductive) +
theme_bw() +
labs(x="Junction RPM, steady-state", y="Junction RPM, naRNA")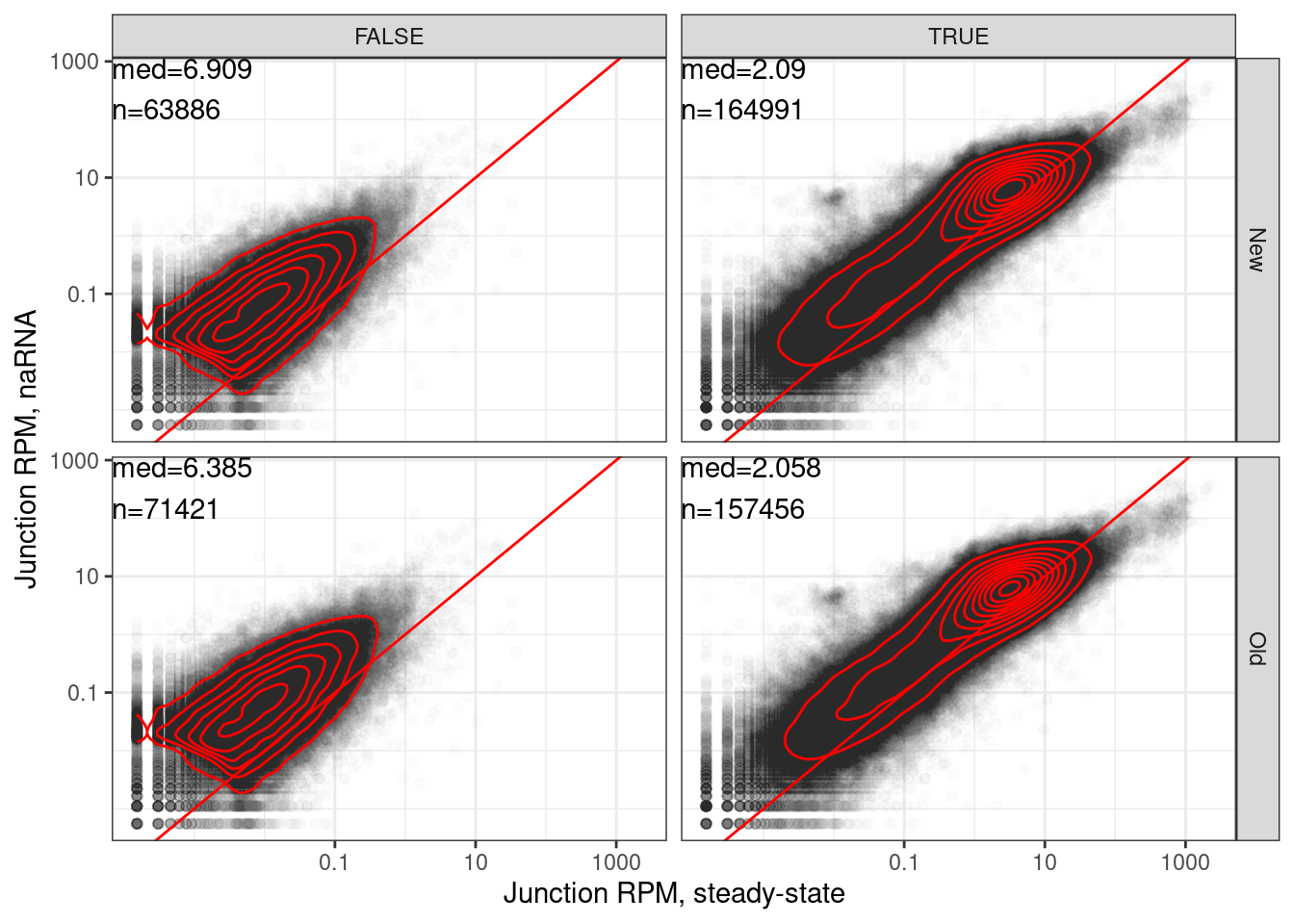
…same plot but just new method
dat.to.Plot <- juncs.long.summary.joined.toplot %>%
mutate(ProductiveOrUnproductive = case_when(
(Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene") ~ "Productive\nSplice juncs",
!(Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene") ~ "Unproductive\nSplice juncs",
))
ggplot(dat.to.Plot, aes(x=Expression.Splicing, y=chRNA.Expression.Splicing)) +
geom_point(alpha=0.01, color='black') +
geom_density2d(color='red') +
geom_abline(slope = 1, color='red') +
geom_text(data = . %>%
replace_na(list(Expression.Splicing=1E-5, chRNA.Expression.Splicing=1E-5)) %>%
group_by(ProductiveOrUnproductive) %>%
summarise(med = round(median(chRNA.Expression.Splicing/Expression.Splicing, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("median FC={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1.1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
facet_wrap(~ProductiveOrUnproductive) +
theme_bw() +
coord_fixed() +
labs(x="Junction RPM, steady-state", y="Junction RPM, naRNA")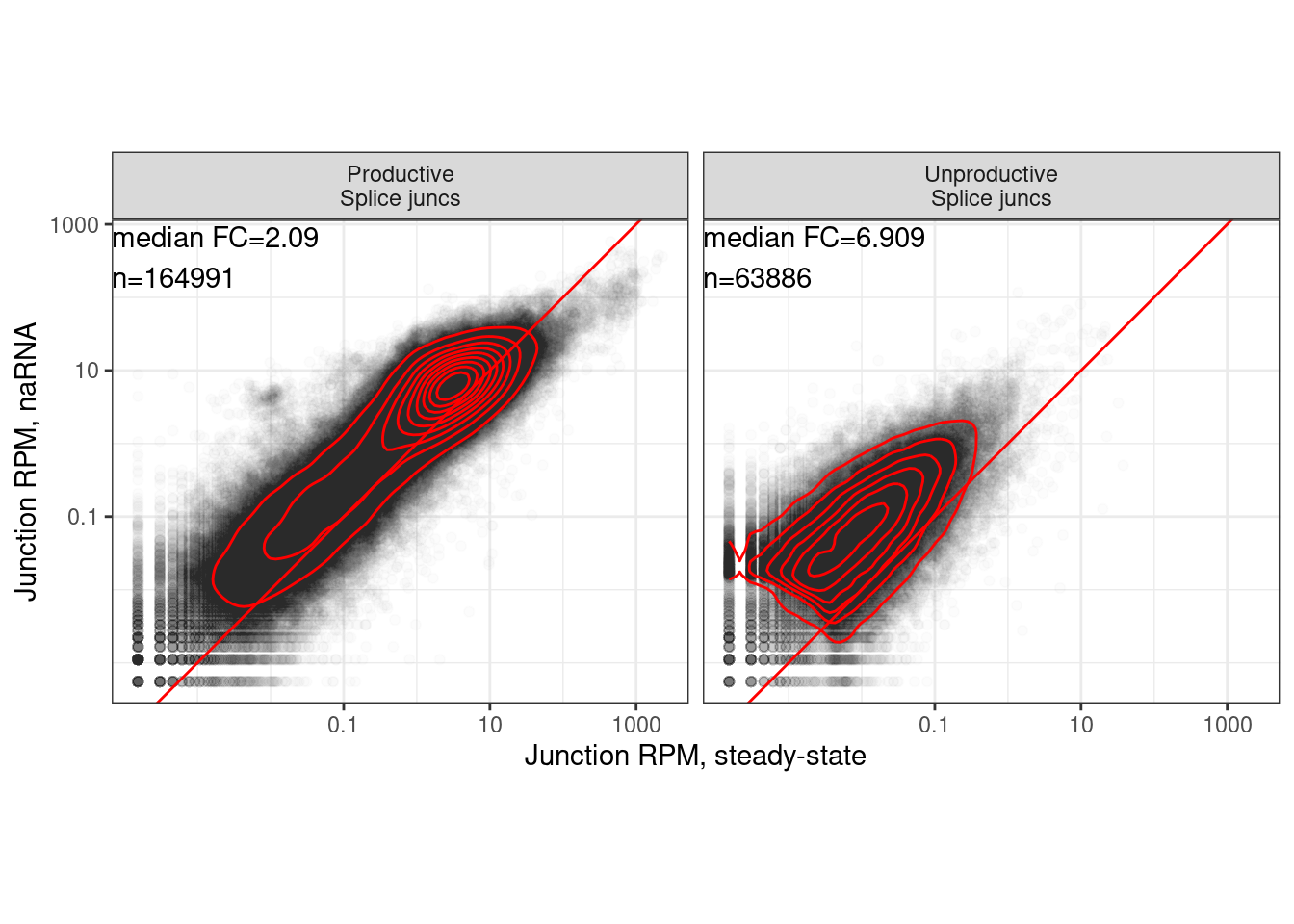
ggsave("../code/scratch/PlotsForYangGrant.JunctionScatter.Points.pdf", height=3, width=7)
ggplot(dat.to.Plot, aes(x=Expression.Splicing, y=chRNA.Expression.Splicing)) +
# geom_point(alpha=0.01, color='black') +
# geom_density2d(color='red') +
geom_hex(bins=70) +
geom_abline(slope = 1, color='red') +
geom_text(data = . %>%
replace_na(list(Expression.Splicing=1E-5, chRNA.Expression.Splicing=1E-5)) %>%
group_by(ProductiveOrUnproductive) %>%
summarise(med = round(median(chRNA.Expression.Splicing/Expression.Splicing, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue(" median FC={med}\n n={n}")),
x=-Inf, y=Inf, vjust=1.1, hjust=0.) +
scale_color_identity() +
scale_fill_viridis_c(trans='log10', option = 'B') +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
facet_wrap(~ProductiveOrUnproductive) +
theme_bw() +
coord_fixed() +
labs(x="Junction RPM, steady-state", y="Junction RPM, naRNA")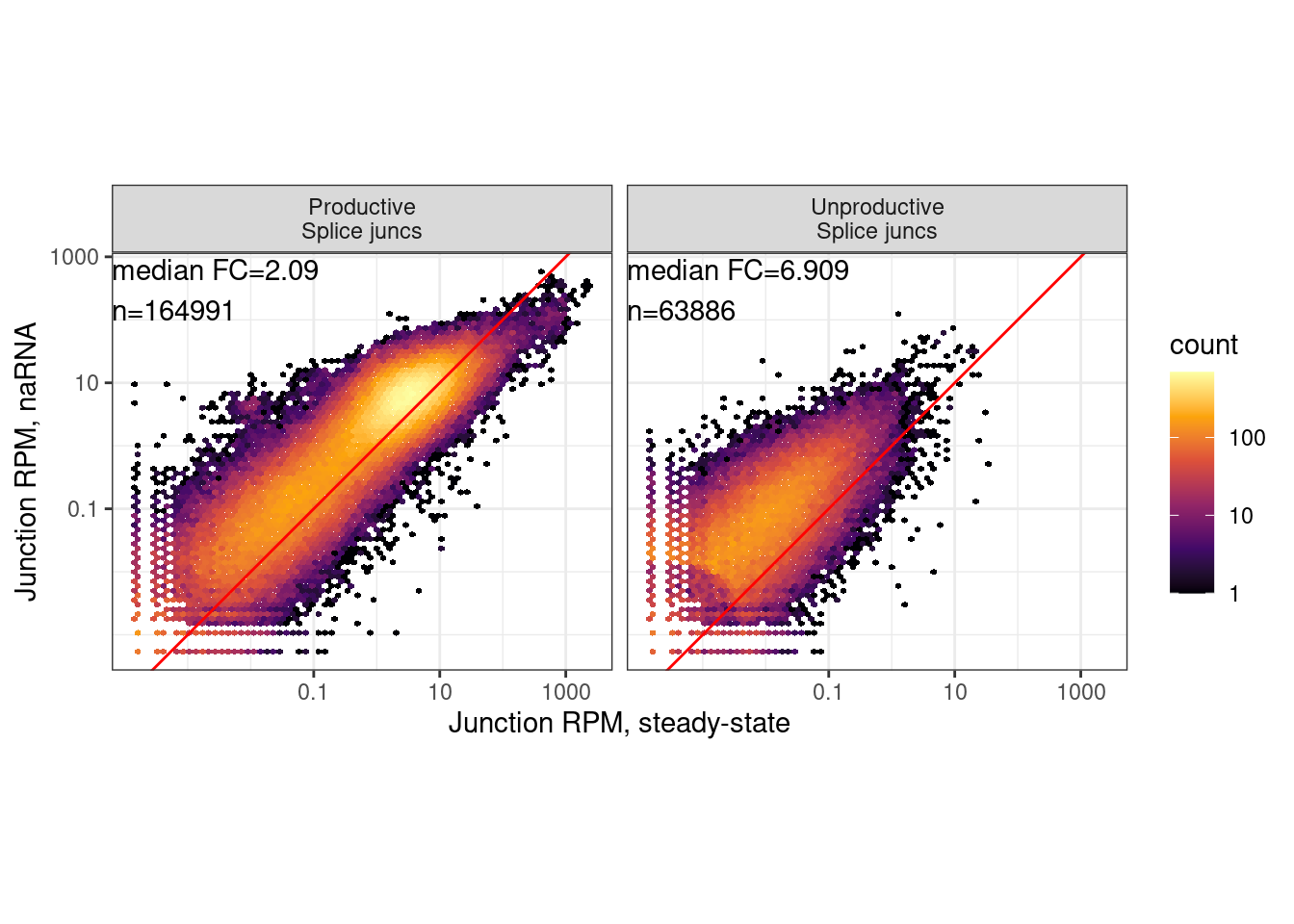
ggsave("../code/scratch/PlotsForYangGrant.JunctionScatter.Hexbin.pdf", height=2, width=4.25)Plots for Yang’s grant.
juncs.long.summary.joined.toplot <- juncs.long.summary.joined %>%
filter(!UTR) %>%
filter(Dataset %in% c("HeLa.scr", "HeLa.dKD")) %>%
dplyr::select(-DatasetSum, -Sum) %>%
pivot_wider(names_from="Dataset", values_from="RPM") %>%
mutate(Color = recode(SuperAnnotation, AnnotatedJunc_NoncodingGene="#6a3d9a", UnannotatedJunc_NoncodingJunc="#cab2d6", AnnotatedJunc_UnproductiveCodingGene="#e31a1c", UnannotatedJunc_UnproductiveCodingGene="#fb9a99", AnnotatedJunc_ProductiveCodingGene="#1f78b4", UnannotatedJunc_ProductiveCodingGene="#a6cee3")) %>%
mutate(SuperAnnotation = factor(SuperAnnotation, levels=c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene", "AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene", "AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))) %>%
filter(!SuperAnnotation %in% c("AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))
bind_rows(
juncs.long.summary.joined.toplot %>%
mutate(method="Old"),
juncs.long.summary.joined.toplot %>%
mutate(method="New"),
) %>%
mutate(ProductiveOrUnproductive = case_when(
method == "New" & (Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene") ~ TRUE,
method == "New" & !(Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene") ~ FALSE,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene") ~ TRUE,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene") ~ FALSE
)) %>%
mutate(ProductiveOrUnproductive = if_else(ProductiveOrUnproductive, "Productive", "Unproductive")) %>%
ggplot(aes(x=HeLa.scr, y=HeLa.dKD)) +
# geom_scattermore(alpha=0.01, color='black') +
# geom_hex() +
# scale_fill_viridis_c()
geom_point(alpha=0.01, color='black') +
geom_density2d(color='red') +
geom_text(data = . %>%
replace_na(list(HeLa.scr=1E-5, HeLa.dKD=1E-5)) %>%
group_by(method, ProductiveOrUnproductive) %>%
summarise(med = round(median(HeLa.dKD/HeLa.scr, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("Med={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
geom_abline(slope = 1, color='red') +
facet_grid(method~ProductiveOrUnproductive) +
theme_classic() +
labs(x="Junction RPM, control", y="Junction RPM, dKD", caption="Filtered out UTR juncs.\nVertical facets are 'Coding' value")
ggsave("/project2/yangili1/bjf79/scratch/EvaluateJuncClassification.shRNAKD.pdf", height=6, width=3)
bind_rows(
juncs.long.summary.joined.toplot %>%
mutate(method="Old"),
juncs.long.summary.joined.toplot %>%
mutate(method="New"),
) %>%
mutate(ProductiveOrUnproductive = case_when(
method == "New" & (Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene") ~ TRUE,
method == "New" & !(Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene") ~ FALSE,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene") ~ TRUE,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene") ~ FALSE
)) %>%
mutate(ProductiveOrUnproductive = if_else(ProductiveOrUnproductive, "Productive", "Unproductive")) %>%
filter(method == "New") %>%
ggplot(aes(x=HeLa.scr, y=HeLa.dKD)) +
geom_point(alpha=0.01, color='black') +
geom_density2d(color='red') +
geom_text(data = . %>%
replace_na(list(HeLa.scr=1E-5, HeLa.dKD=1E-5)) %>%
group_by(ProductiveOrUnproductive) %>%
summarise(med = round(median(HeLa.dKD/HeLa.scr, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("Med={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
geom_abline(slope = 1, color='red') +
facet_wrap(~ProductiveOrUnproductive) +
theme_classic() +
labs(x="Junction RPM, control", y="Junction RPM, dKD", caption="Filtered out UTR juncs.\nVertical facets are 'Coding' value")
# ggsave("/project2/yangili1/bjf79/scratch/EvaluateJuncClassification.shRNAKD.NoOld.pdf", height=3, width=3)
juncs.long.summary.joined.toplot <- juncs.long.summary.joined %>%
filter(!UTR) %>%
filter(Dataset %in% c("Expression.Splicing", "chRNA.Expression.Splicing")) %>%
dplyr::select(-DatasetSum, -Sum) %>%
pivot_wider(names_from="Dataset", values_from="RPM") %>%
mutate(Color = recode(SuperAnnotation, AnnotatedJunc_NoncodingGene="#6a3d9a", UnannotatedJunc_NoncodingJunc="#cab2d6", AnnotatedJunc_UnproductiveCodingGene="#e31a1c", UnannotatedJunc_UnproductiveCodingGene="#fb9a99", AnnotatedJunc_ProductiveCodingGene="#1f78b4", UnannotatedJunc_ProductiveCodingGene="#a6cee3")) %>%
mutate(SuperAnnotation = factor(SuperAnnotation, levels=c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene", "AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene", "AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))) %>%
filter(!SuperAnnotation %in% c("AnnotatedJunc_NoncodingGene", "UnannotatedJunc_NoncodingJunc"))
bind_rows(
juncs.long.summary.joined.toplot %>%
mutate(method="Old"),
juncs.long.summary.joined.toplot %>%
mutate(method="New"),
) %>%
mutate(ProductiveOrUnproductive = case_when(
method == "New" & (Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene") ~ TRUE,
method == "New" & !(Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene") ~ FALSE,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene") ~ TRUE,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene") ~ FALSE
)) %>%
mutate(ProductiveOrUnproductive = if_else(ProductiveOrUnproductive, "Productive", "Unproductive")) %>%
ggplot(aes(x=Expression.Splicing, y=chRNA.Expression.Splicing)) +
geom_point(alpha=0.01, color='black') +
geom_density2d(color='red') +
geom_text(data = . %>%
replace_na(list(Expression.Splicing=1E-5, chRNA.Expression.Splicing=1E-5)) %>%
group_by(method, ProductiveOrUnproductive) %>%
summarise(med = round(median(chRNA.Expression.Splicing/Expression.Splicing, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("Med={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
geom_abline(slope = 1, color='red') +
facet_grid(method~ProductiveOrUnproductive) +
theme_classic() +
labs(x="Junction RPM, steady-state", y="Junction RPM, naRNA", caption="Filtered out UTR juncs.\nVertical facets are 'Coding' value")
# ggsave("/project2/yangili1/bjf79/scratch/EvaluateJuncClassification.naRNA.pdf", height=6, width=3)
bind_rows(
juncs.long.summary.joined.toplot %>%
mutate(method="Old"),
juncs.long.summary.joined.toplot %>%
mutate(method="New"),
) %>%
mutate(ProductiveOrUnproductive = case_when(
method == "New" & (Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene") ~ TRUE,
method == "New" & !(Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene") ~ FALSE,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_ProductiveCodingGene", "UnannotatedJunc_ProductiveCodingGene") ~ TRUE,
method == "Old" & SuperAnnotation %in% c("AnnotatedJunc_UnproductiveCodingGene", "UnannotatedJunc_UnproductiveCodingGene") ~ FALSE
)) %>%
mutate(ProductiveOrUnproductive = if_else(ProductiveOrUnproductive, "Productive", "Unproductive")) %>%
filter(method == "New") %>%
ggplot(aes(x=Expression.Splicing, y=chRNA.Expression.Splicing)) +
geom_point(alpha=0.01, color='black') +
geom_density2d(color='red') +
geom_text(data = . %>%
replace_na(list(Expression.Splicing=1E-5, chRNA.Expression.Splicing=1E-5)) %>%
group_by(ProductiveOrUnproductive) %>%
summarise(med = round(median(chRNA.Expression.Splicing/Expression.Splicing, na.rm=F), 3),
n = n()) %>%
ungroup(),
aes(label=str_glue("Med={med}\nn={n}")),
x=-Inf, y=Inf, vjust=1, hjust=0.) +
scale_color_identity() +
scale_x_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
scale_y_continuous(trans="log10", breaks=c(1E-1, 10, 1000), labels=c("0.1", "10", "1000")) +
geom_abline(slope = 1, color='red') +
facet_wrap(~ProductiveOrUnproductive) +
theme_classic() +
labs(x="Junction RPM, steady-state", y="Junction RPM, naRNA", caption="Filtered out UTR juncs.\nVertical facets are 'Coding' value")
# ggsave("/project2/yangili1/bjf79/scratch/EvaluateJuncClassification.naRNA.NoOld.pdf", height=3, width=3)Test updated script
Yang updated his script to reflect this new change… I reran the script and now let’s verify the new annotations.
NewScript.Annotations <- read_tsv("/project/yangili1/bjf79/2024_NMD_Junction_Classifier/Leaf2_updated_junction_classifications.txt") %>%
separate(Intron_coord, into=c("chrom", "start", "end"), sep="[:-]", convert=T, remove = F) %>%
add_count(Intron_coord, name="NumberOfEntriesWithSameCoords")
juncs.long.summary.joined.toplot %>%
inner_join(
NewScript.Annotations %>%
dplyr::select(Intron_coord, CodingUpdated = Coding)
) %>%
mutate(ProductiveOrUnproductiveMyTest = case_when(
Coding | SuperAnnotation=="AnnotatedJunc_ProductiveCodingGene" ~ TRUE,
TRUE ~ FALSE)) %>%
count(CodingUpdated, ProductiveOrUnproductiveMyTest)# A tibble: 3 × 3
CodingUpdated ProductiveOrUnproductiveMyTest n
<lgl> <lgl> <int>
1 FALSE FALSE 63886
2 FALSE TRUE 11
3 TRUE TRUE 164980The script is correctly updated, such that the Coding column in the new srcipt output is true if it can create a transcript that reaches a stop codon, OR if it is already in an annotated functional transcript.
Write organized metadata to help Carlos upload data to GEO
Metadata <- read_tsv("../code/config/samples.tsv")
Metadata %>%
filter(Phenotype == "H3K36ME3") %>%
distinct(IndID) %>%
inner_join(
read_tsv("/project2/yangili1/bjf79/ChromatinSplicingQTLs/data/igsr_samples.tsv.gz"),
by=c("IndID"="Sample name")
) %>%
count(Sex)
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.3.13-el7-x86_64/lib/libopenblas_haswellp-r0.3.13.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=C
[4] LC_COLLATE=C LC_MONETARY=C LC_MESSAGES=C
[7] LC_PAPER=C LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] scattermore_0.8 data.table_1.14.2 forcats_0.5.1 stringr_1.4.0
[5] dplyr_1.0.9 purrr_0.3.4 readr_2.1.2 tidyr_1.2.0
[9] tibble_3.1.7 ggplot2_3.3.6 tidyverse_1.3.1
loaded via a namespace (and not attached):
[1] fs_1.5.2 lubridate_1.8.0 bit64_4.0.5 httr_1.4.3
[5] rprojroot_2.0.3 tools_4.2.0 backports_1.4.1 bslib_0.3.1
[9] utf8_1.2.2 R6_2.5.1 DBI_1.1.2 colorspace_2.0-3
[13] withr_2.5.0 tidyselect_1.1.2 bit_4.0.4 compiler_4.2.0
[17] git2r_0.30.1 textshaping_0.3.6 cli_3.3.0 rvest_1.0.2
[21] xml2_1.3.3 isoband_0.2.5 labeling_0.4.2 sass_0.4.1
[25] scales_1.2.0 hexbin_1.28.3 systemfonts_1.0.4 digest_0.6.29
[29] rmarkdown_2.14 R.utils_2.11.0 pkgconfig_2.0.3 htmltools_0.5.2
[33] dbplyr_2.1.1 fastmap_1.1.0 highr_0.9 rlang_1.0.2
[37] readxl_1.4.0 rstudioapi_0.13 jquerylib_0.1.4 farver_2.1.0
[41] generics_0.1.2 jsonlite_1.8.0 vroom_1.5.7 R.oo_1.24.0
[45] magrittr_2.0.3 Rcpp_1.0.8.3 munsell_0.5.0 fansi_1.0.3
[49] lifecycle_1.0.1 R.methodsS3_1.8.1 stringi_1.7.6 whisker_0.4
[53] yaml_2.3.5 MASS_7.3-56 grid_4.2.0 parallel_4.2.0
[57] promises_1.2.0.1 crayon_1.5.1 lattice_0.20-45 haven_2.5.0
[61] hms_1.1.1 knitr_1.39 pillar_1.7.0 reprex_2.0.1
[65] glue_1.6.2 evaluate_0.15 modelr_0.1.8 vctrs_0.4.1
[69] tzdb_0.3.0 httpuv_1.6.5 cellranger_1.1.0 gtable_0.3.0
[73] assertthat_0.2.1 xfun_0.30 broom_0.8.0 later_1.3.0
[77] ragg_1.2.5 viridisLite_0.4.0 workflowr_1.7.0 ellipsis_0.3.2